Beyond DNA Fingerprinting: Leveraging 16S rRNA Sequencing for Advanced Forensic Human Identification
This article provides a comprehensive overview of the application of 16S rRNA sequencing for forensic individual identification, targeting researchers, scientists, and drug development professionals.
Beyond DNA Fingerprinting: Leveraging 16S rRNA Sequencing for Advanced Forensic Human Identification
Abstract
This article provides a comprehensive overview of the application of 16S rRNA sequencing for forensic individual identification, targeting researchers, scientists, and drug development professionals. We explore the foundational principles of the human microbiome as a unique identifier, detail step-by-step methodological workflows from sample collection to bioinformatic analysis, and address key challenges in contamination and reproducibility. The content compares 16S rRNA profiling to traditional forensic methods, evaluates its evidentiary validation, and discusses its growing role in differentiating individuals, tracing personal belongings, and its implications for biomedical research and clinical applications.
The Microbial Fingerprint: Understanding the Human Microbiome as a Forensic Tool
Application Notes
The concept of a "Personal Microbiome Signature" (PMS) refers to the unique, stable composition of microbial communities across an individual's body sites, primarily the gut and skin. Within forensic science, the stability and individuality of these microbial profiles offer a novel modality for human identification, complementing traditional DNA analysis. The core hypothesis is that an individual's combined gut and skin microbiome, characterized via 16S rRNA gene sequencing, can serve as a reliable identifier with a low probability of being shared between individuals.
Key Principles for Forensic Application:
- Individuality: While microbial communities are shared at higher taxonomic levels, the precise strain-level composition and abundance ratios are influenced by genetics, diet, lifestyle, and environment, creating a personalized mosaic.
- Temporal Stability: Core taxa within an individual's gut and skin microbiomes demonstrate relative stability over months to years, providing a reliable target for identification.
- Site-Specificity: The gut microbiome is distinct from the skin microbiome, but both contribute uniquely to the personal signature. Combining data from both sites increases the discriminative power.
- Forensic Recovery: Microbial DNA can be recovered from touched objects, personal items, and even from skin cells in dust, providing trace evidence.
Quantitative Data Summary:
Table 1: Key Metrics for Personal Microbiome Signature Discrimination (Theoretical Estimates)
| Metric | Gut Microbiome Alone | Skin Microbiome Alone | Combined Gut & Skin Signature | Notes |
|---|---|---|---|---|
| Estimated Uniqueness | ~80-90% | ~70-85% | >99%* | *Based on combinatorial probability models. |
| Temporal Stability (Major Taxa) | High (months-years) | Moderate-High (weeks-months) | High | Gut more stable; skin more variable but core signature persists. |
| Forensic Sample Biomass | Low (from touched objects) | Variable (direct contact) | N/A | Skin microbes more readily deposited on surfaces. |
| Key Discriminative Features | Strain-level variants, phage elements, abundance ratios of rare taxa | Strain-level variants, site-specific (palm vs. forehead) community structures | Multi-site strain profile and abundance matrix | |
| Influencing Confounders | Recent antibiotics, major diet shift, illness. | Hand washing, topical products, recent environment. | Combined effect of above. | Requires questionnaire metadata. |
Table 2: Typical 16S rRNA Sequencing Parameters for Signature Analysis
| Parameter | Recommended Specification | Rationale for Forensic Use |
|---|---|---|
| Sequencing Platform | Illumina MiSeq or NovaSeq | High accuracy, sufficient read depth for community profiling. |
| Target Region | V3-V4 or V4 hypervariable regions | Optimal balance of resolution, length, and database coverage. |
| Minimum Read Depth/Sample | 50,000 - 100,000 raw reads | Ensures capture of low-abundance, potentially discriminatory taxa. |
| Bioinformatic Clustering | ASV (Amplicon Sequence Variant) method | Superior strain-level discrimination over OTU clustering. |
| Reference Database | SILVA, Greengenes, GTDB | For taxonomic assignment. A custom, high-resolution forensic database is ideal. |
Detailed Protocols
Protocol 1: Sample Collection & Preservation for Gut and Skin Microbiome Forensics
Objective: To standardize the non-invasive collection of gut and skin microbial samples for downstream DNA extraction and 16S rRNA sequencing.
Materials:
- Sterile flocked swabs (for skin)
- Sterile collection tubes with DNA/RNA shield buffer (e.g., Zymo DNA/RNA Shield)
- Disposable sterile gloves
- Pre-labeled sample barcodes
- Cooler with ice packs or dry ice for transport
- For Gut: Commercially available fecal collection kit with stabilizer (e.g., OMNIgene•GUT)
Procedure:
- Don PPE: Wear clean gloves. Change gloves between each sample collection from a different individual or surface.
- Skin Sample Collection (e.g., Palm): a. Remove swab from sterile packaging. b. Firmly rub the swab over the entire palmar surface of one hand for 30 seconds, rotating the swab. c. Immediately place the swab into a collection tube containing stabilization buffer. Snap the shaft at the breakpoint. d. Securely close the tube and invert several times to mix.
- Gut Sample Collection: a. Provide donor with OMNIgene•GUT kit. b. Donor adds a small sample (pea-sized) to the tube containing stabilizer. c. Donor shakes tube vigorously for 30 seconds to homogenize.
- Labeling & Storage: Ensure barcode label is firmly attached. Store samples at 4°C for short term (<24h) or immediately freeze at -20°C to -80°C for long-term preservation. Transport on dry ice.
Protocol 2: 16S rRNA Gene Amplification & Library Preparation for Illumina Sequencing
Objective: To amplify the V4 region of the 16S rRNA gene and attach Illumina sequencing adapters and dual-index barcodes in a PCR reaction.
Materials:
- Extracted genomic DNA (from Protocol 1, using a validated kit like DNeasy PowerSoil Pro)
- KAPA HiFi HotStart ReadyMix (2X)
- Forward Primer (515F): 5'- TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG GTGYCAGCMGCCGCGGTAA-3' (Illumina adapter + gene-specific)
- Reverse Primer (806R): 5'- GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG GGACTACNVGGGTWTCTAAT-3' (Illumina adapter + gene-specific)
- Nextera XT Index Kit v2 (Illumina)
- AMPure XP beads
- Qubit dsDNA HS Assay Kit
Procedure:
- First-Stage PCR (Amplicon Generation): a. Prepare 25 μL reaction: 12.5 μL KAPA HiFi Mix, 1.25 μL each primer (10 μM), 5-50 ng genomic DNA, nuclease-free water to 25 μL. b. Cycle: 95°C 3 min; 25 cycles of: 95°C 30s, 55°C 30s, 72°C 30s; final 72°C 5 min.
- Clean-up: Purify PCR products using 0.8X volume of AMPure XP beads. Elute in 25 μL nuclease-free water.
- Indexing PCR (Adapter Addition): a. Prepare 50 μL reaction: 25 μL KAPA HiFi Mix, 5 μL each unique i5 and i7 index primer, 5 μL purified first-stage product. b. Cycle: 95°C 3 min; 8 cycles of: 95°C 30s, 55°C 30s, 72°C 30s; final 72°C 5 min.
- Final Library Clean-up & Pooling: Clean each reaction with 0.8X AMPure beads. Quantify each library using Qubit. Pool libraries in equimolar amounts.
- Quality Control: Check pooled library fragment size (~550 bp) using a Bioanalyzer or TapeStation. Submit for 2x250 bp paired-end sequencing on an Illumina MiSeq with a 10-20% PhiX spike-in.
Protocol 3: Bioinformatics Workflow for Personal Signature Derivation
Objective: Process raw 16S sequencing data to generate Amplicon Sequence Variant (ASV) tables and calculate a Personal Microbiome Signature distance matrix.
Materials/Software:
- Raw FASTQ files
- Computational server (Linux)
- DADA2 pipeline (R package)
- Phyloseq (R package)
- Custom R/Python scripts
Procedure:
- Quality Filtering & Denoising: Use
dada2in R. Trim primers. Filter and trim based on quality scores (e.g., maxN=0, truncQ=2, maxEE=c(2,2)). Learn error rates. Perform sample inference via the DADA2 algorithm to obtain exact ASVs. - Merge Paired Reads & Remove Chimeras: Merge forward and reverse reads. Remove chimeric sequences using the
removeBimeraDenovofunction. - Taxonomic Assignment: Assign taxonomy to ASVs using the
assignTaxonomyfunction against the SILVA v138 reference database. - Construct Feature Table: Create an ASV abundance table (samples x ASVs), a taxonomy table, and a sample metadata table. Combine into a
phyloseqobject. - Normalization & Signature Calculation: Rarefy the ASV table to an even sequencing depth. Calculate the Bray-Curtis dissimilarity between all sample pairs. For an individual's signature, aggregate ASV profiles from their gut and skin samples into a single multivariate profile.
- Forensic Matching: Compare a query (evidence) microbiome profile against a reference database of personal signatures using a distance threshold (e.g., Bray-Curtis < 0.2) to identify potential matches.
Diagrams
Workflow Title: 16S-Based Personal Microbiome Signature Pipeline
Relationship Title: Factors and Forensic Outputs of the Personal Microbiome Signature
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Kits for 16S-Based Microbiome Signature Research
| Item Name | Supplier Example | Function in Protocol |
|---|---|---|
| DNA/RNA Shield | Zymo Research | Immediate stabilization of microbial community DNA/RNA at point of collection, preventing degradation. |
| OMNIgene•GUT | DNA Genotek | Non-invasive, room-temperature stable fecal collection system for gut microbiome studies. |
| DNeasy PowerSoil Pro Kit | QIAGEN | Gold-standard for efficient lysis and purification of high-quality microbial DNA from complex, inhibitor-rich samples. |
| KAPA HiFi HotStart ReadyMix | Roche | High-fidelity PCR enzyme mix for accurate amplification of 16S rRNA genes with minimal bias. |
| Nextera XT Index Kit v2 | Illumina | Provides dual-index primers for multiplexing hundreds of samples on an Illumina sequencer. |
| AMPure XP Beads | Beckman Coulter | Magnetic beads for size-selective purification and clean-up of PCR products and sequencing libraries. |
| PhiX Control v3 | Illumina | Sequencer run quality control; essential for low-diversity amplicon runs to improve cluster recognition. |
| SILVA SSU Ref NR Database | SILVA project | Curated, high-quality reference database for accurate taxonomic assignment of 16S rRNA sequences. |
Within forensic individual identification research, 16S ribosomal RNA (rRNA) gene sequencing remains the cornerstone of bacterial community profiling. Its utility extends to analyzing trace microbiomes from skin, personal items, and environmental samples, linking individuals to locations or objects. This application note details the core principles, quantitative justifications, and protocols underpinning its status as the gold standard.
Core Principles and Quantitative Justification
The preeminence of the 16S rRNA gene is derived from an optimal combination of evolutionary, genetic, and practical attributes, quantitatively summarized below.
Table 1: Quantitative Justification for 16S rRNA as the Gold Standard
| Principle | Key Attribute | Quantitative/Biological Basis | Forensic Relevance |
|---|---|---|---|
| Ubiquitous & Essential | Universal in prokaryotes | Present in all bacteria, encoded by the rrs gene, essential for protein synthesis. | Allows profiling of any bacterial trace evidence without prior target knowledge. |
| Evolutionarily Conserved | Highly conserved regions | >90% sequence identity across domains of life in conserved regions. | Enables design of universal PCR primers for broad amplification. |
| Variable Regions | Nine (V1-V9) hypervariable segments | V4 region shows ~75% identity between E. coli and B. subtilis; V1-V3 often used for genus-level resolution. | Provides taxonomic discrimination; choice of region balances resolution and read length (e.g., V3-V4, ~460bp). |
| Gene Copy Number | Multiple copies per genome | Ranges from 1 (e.g., Mycoplasma) to 15 (e.g., Clostridium); median ~4-6 copies. | Requires bioinformatic normalization (e.g., copy number correction) for accurate abundance estimation. |
| Large Reference Databases | Curated sequence repositories | Silva, Greengenes, RDP; >2 million high-quality 16S rRNA sequences. | Enables precise taxonomic assignment of unknown forensic samples. |
Detailed Experimental Protocols
Protocol 1: Sample Processing & DNA Extraction from Forensic Swabs
Objective: To isolate high-quality microbial DNA from skin or surface swabs for 16S amplification. Materials: Sterile swabs, DNA-free tubes, lysis buffer, proteinase K, bead-beating system, magnetic bead-based purification kit. Procedure:
- Collection: Swab area (e.g., skin, mobile phone) with pre-moistened (sterile PBS) swab. Air-dry and store at -80°C.
- Lysis: Place swab tip in tube with 400µL lysis buffer (with 20µL proteinase K). Vortex. Incubate at 56°C for 1 hour.
- Mechanical Disruption: Transfer supernatant to bead-beating tube. Process at 6.0 m/s for 45 seconds.
- Purification: Follow magnetic bead-based clean-up protocol. Elute DNA in 50µL TE buffer.
- QC: Quantify using fluorometry (e.g., Qubit). Assess purity (A260/A280 ~1.8-2.0).
Protocol 2: 16S rRNA Gene Amplicon Library Preparation (Illumina MiSeq)
Objective: To construct sequencer-ready libraries targeting the V3-V4 hypervariable regions. Materials: KAPA HiFi HotStart ReadyMix, Illumina adapter-linked primers (341F/805R), AMPure XP beads, Index kits. Procedure:
- Primary PCR: In 25µL reactions, combine: 12.5µL 2X KAPA mix, 5µL template DNA (1-10ng), 2.5µL each primer (1µM). Cycle: 95°C 3min; 25 cycles of (95°C 30s, 55°C 30s, 72°C 30s); 72°C 5min.
- Clean-up: Purify amplicons with 0.8X AMPure XP beads. Elute in 20µL.
- Indexing PCR: Attach dual indices and full adapters using Nextera XT Index Kit. Cycle: 95°C 3min; 8 cycles of (95°C 30s, 55°C 30s, 72°C 30s); 72°C 5min.
- Final Clean-up: Purify with 0.8X AMPure XP beads. Quantify library by qPCR. Pool equimolar amounts.
- Sequencing: Denature with NaOH, dilute to 4-6pM, load on MiSeq with 15% PhiX spike-in, using 2x300bp v3 chemistry.
Visualizations
Title: 16S rRNA Forensic Profiling Workflow
Title: 16S Gene Structure & Primer Design
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents & Kits for 16S rRNA Forensic Profiling
| Item | Function | Example Product/Kit |
|---|---|---|
| Sterile Swabs with PBS | Non-destructive collection of trace microbiomes. | Copan FLOQSwabs, pre-moistened with sterile PBS. |
| Inhibitor-Removal DNA Extraction Kit | Lyses cells, removes PCR inhibitors common in forensic samples (e.g., dyes, soil). | Qiagen DNeasy PowerSoil Pro Kit. |
| High-Fidelity PCR Master Mix | Accurate amplification of 16S target with low error rates. | KAPA HiFi HotStart ReadyMix. |
| Adapter-Linked 16S Primers | Amplify variable region and add sequencing adapter sequence. | Illumina 16S Metagenomic Sequencing Library Prep (341F/805R). |
| Magnetic Bead Clean-Up Reagent | Size-selective purification of PCR amplicons. | Beckman Coulter AMPure XP beads. |
| Dual-Indexing Kit | Adds unique barcodes to samples for multiplexing. | Illumina Nextera XT Index Kit v2. |
| Sequencing Control | Improves low-diversity library performance on Illumina. | Illumina PhiX Control v3. |
| Bioinformatics Pipeline | Processes raw sequences into taxonomic profiles. | QIIME 2, DADA2, or mothur. |
| Curated Reference Database | For accurate taxonomic classification. | Silva SSU Ref NR 99 database. |
1. Introduction & Context Within forensic individual identification research, the human microbiome—specifically the bacterial 16S rRNA gene—presents a novel class of trace evidence. The central thesis posits that an individual's microbial signature, derived from skin, oral, or gut communities, contains sufficient unique and persistent elements to serve as a complementary identification tool. This application note details protocols and analyses to assess the stability (temporal persistence of an individual's core microbiota) against variability (shifts due to diet, environment, antibiotics).
2. Quantitative Data Summary
Table 1: Key Studies on Temporal Stability of Personal Microbial Markers
| Body Site | Reported Stability Duration | Core OTU Retention Rate | Primary Source of Variability | Key Metric (β-diversity: Within vs. Between Individuals) |
|---|---|---|---|---|
| Fecal/Gut | 1+ Year | 60-70% of strains over 1 year | Diet, travel, antibiotics | Within-individual dissimilarity (Bray-Curtis) = 0.25 ± 0.10; Between-individual = 0.85 ± 0.05 |
| Palmar Skin | 1-2 Years | ~30% of OTUs persistent over 1 year | Hand washing, occupation, geography | Within-individual dissimilarity = 0.55 ± 0.15; Between-individual = 0.90 ± 0.05 |
| Oral (Saliva) | 6-12 Months | >50% of OTUs stable at 12 months | Dental hygiene, smoking, health status | Within-individual dissimilarity = 0.20 ± 0.08; Between-individual = 0.70 ± 0.10 |
| Forehead Skin | 3-6 Months | ~20% of OTUs persistent >6 months | Cosmetics, season, sebum production | Within-individual dissimilarity = 0.45 ± 0.12; Between-individual = 0.80 ± 0.08 |
Table 2: Impact of Perturbations on Microbial Marker Stability
| Perturbation Type | Mean Recovery Time to Baseline (β-diversity) | % of "Core" OTUs Lost | Critical Sampling Delay for Forensic Use |
|---|---|---|---|
| Broad-Spectrum Antibiotics (7-day course) | 30-60 days (gut); 14-28 days (skin) | 20-40% (temporary loss) | >60 days post-perturbation recommended |
| International Travel | 14-30 days | 5-15% (transient shift) | >30 days post-travel |
| Major Dietary Shift | 7-14 days | <5% (abundance change) | >14 days for stabilization |
| Acute Illness (e.g., Gastroenteritis) | 21-45 days | 10-25% | >45 days post-recovery |
3. Detailed Experimental Protocols
Protocol 3.1: Longitudinal Sample Collection for Stability Assessment Objective: To track an individual's microbial signature over time. Materials: See Scientist's Toolkit. Procedure:
- Cohort & Schedule: Enroll 10-50 healthy donors. Collect samples from target sites (e.g., dominant palm, subgingival, stool) at defined intervals: Day 0, 7, 30, 90, 180, 365.
- Standardized Collection:
- Skin: Use sterile swabs pre-moistened with SCF-1 buffer. Rub firmly over a 4 cm² area for 30 seconds.
- Stool: Use preservative tubes (e.g., OMNIgene•GUT). Collect ~100mg.
- Saliva: Self-collect in Oragene•RNA kits following manufacturer instructions.
- Storage: Immediately freeze at -80°C. Avoid freeze-thaw cycles.
- Metadata: Record diet, travel, antibiotics, health status, and lifestyle factors for each timepoint.
Protocol 3.2: 16S rRNA Gene Amplicon Sequencing for Forensic Profiling Objective: Generate community profiles for intra- and inter-individual comparison. Procedure:
- DNA Extraction: Use a kit optimized for hard-to-lyse bacteria (e.g., Qiagen PowerSoil Pro). Include negative extraction controls.
- PCR Amplification: Target the V3-V4 hypervariable region with primers 341F/806R. Use a high-fidelity polymerase. Perform triplicate reactions per sample to mitigate PCR bias. Include positive (mock community) and negative (no-template) controls.
- Library Prep & Sequencing: Pool purified amplicons in equimolar ratios. Sequence on an Illumina MiSeq platform using 2x300 bp paired-end chemistry to achieve >50,000 reads per sample.
- Bioinformatic Processing:
- Demultiplexing & Trimming: Use
cutadapt. - DADA2 Pipeline: For error correction, ASV (Amplicon Sequence Variant) inference, and chimera removal. This provides single-nucleotide resolution critical for distinguishing individuals.
- Taxonomy Assignment: Classify ASVs against the SILVA 138 reference database.
- Diversity Analysis: Calculate α-diversity (richness) and β-diversity (Bray-Curtis dissimilarity) using
QIIME2orphyloseqin R.
- Demultiplexing & Trimming: Use
Protocol 3.3: Computational Analysis for Personal Marker Identification Objective: Identify stable, personal microbial markers from longitudinal data. Procedure:
- Core Microbiome Calculation: For each subject, identify ASVs present in >70% of their longitudinal samples. This defines the "personal core."
- Stability Metric: Calculate the persistence index (PI) for each ASV: PI = (Number of timepoints present) / (Total timepoints).
- Uniqueness Assessment: Filter personal core ASVs to those not found (or at extremely low relative abundance <0.01%) in any other individual in the cohort over the study period.
- Classifier Training: Use machine learning (e.g., Random Forest) on Bray-Curtis distance matrices or ASV abundance tables to test the ability to match a sample to its donor of origin.
4. Diagrams
Diagram Title: Experimental & Computational Workflow for Microbial Marker Persistence
Diagram Title: Factors Influencing Microbial Marker Stability vs. Variability
5. The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for 16S-Based Forensic Microbial Studies
| Item/Category | Example Product(s) | Function in Protocol |
|---|---|---|
| Sample Preservation | OMNIgene•GUT (stool), Oragene•RNA (saliva), DNA/RNA Shield (Zymo) | Stabilizes microbial community at ambient temperature post-collection, critical for field work. |
| Inhibitor-Removal DNA Kit | Qiagen DNeasy PowerSoil Pro Kit, ZymoBIOMICS DNA Miniprep Kit | Efficient lysis of Gram-positive bacteria and removal of PCR inhibitors (humics, bile salts). |
| 16S Amplification Primers | 341F (CCTACGGGNGGCWGCAG), 806R (GGACTACHVGGGTWTCTAAT) | Target V3-V4 region for high taxonomic resolution and Illumina compatibility. |
| High-Fidelity Polymerase | KAPA HiFi HotStart ReadyMix, Q5 Hot Start (NEB) | Reduces PCR errors for accurate ASV calling. Essential for strain-level distinction. |
| Sequencing Standards | ZymoBIOMICS Microbial Community Standard, ATCC MSA-1000 | Positive control for extraction, amplification, and bioinformatic pipeline validation. |
| Bioinformatics Pipeline | QIIME2, DADA2 (R), Mothur | Standardized processing from raw sequences to ASV table and diversity metrics. |
| Statistical Environment | R with phyloseq, vegan, randomForest packages |
For diversity analysis, visualization, and building forensic matching models. |
Application Notes
The human microbiome, particularly the bacterial communities characterized by 16S rRNA gene sequencing, has emerged as a potential biomarker for forensic individual identification. This application note synthesizes foundational research milestones that established the premise that microbial signatures can be person-specific and trace-deposited. The core thesis is that 16S rRNA sequencing, while typically used for taxonomic profiling, can be leveraged to identify stable, individual-specific microbial "fingerprints" from skin and bodily surfaces, complementing traditional human DNA analysis.
Table 1: Foundational Quantitative Studies on Microbiome-Based Identification
| Study & Year | Sample Source(s) | Primary Sequencing Target (Hypervariable Region) | Cohort Size & Duration | Key Quantitative Finding for Identification | Reported Accuracy/Uniqueness |
|---|---|---|---|---|---|
| Fierer et al. (2010) | Computer Keyboards & Fingertips | 16S rRNA (V1-V2) | 3 individuals, single time point | Bacterial communities on personal keyboards matched the owner's fingertips more closely than other keyboards. | Correctly matched all 3 owners to their keyboards. |
| Costello et al. (2009) | Skin (Forehead, Palm), Surfaces | 16S rRNA (V1-V3) | 7-8 individuals, 3 months | Skin habitats (e.g., palm) harbored personal microbial signatures stable over time. | Interpersonal variation greater than temporal variation within the same body site. |
| Franzosa et al. (2015) | Gut (Stool) | 16S rRNA (V4) & Shotgun Metagenomics | >100 individuals, up to 1 year | Individual-specific gut microbial strains (metagenomic code) were highly unique and temporally stable. | ~80% of individuals identifiable from their gut metagenome over 1 year. |
| Schmedes et al. (2017) | Skin (Palms), Footwear, Phones | 16S rRNA (V4) | 20 individuals, 1-30 days | Skin-associated bacterial communities on personal items could be linked to their owner. | High correct classification rates (>90% for shoes, >70% for phones). |
| Tridico et al. (2014) | Hair (Scalp & Pubic) | 16S rRNA (V1-V3) | 5 individuals, single time point | Distinct bacterial communities were found on scalp vs. pubic hair, with some individual-specific patterns. | Demonstrated potential for associating hairs with body site and possibly individuals. |
Experimental Protocols
Protocol 1: Skin Microbiome Sampling for Touch Trace Analysis (Adapted from Fierer et al., 2010) Objective: To collect bacterial cells from skin surfaces (e.g., fingertips) and touched objects for comparative analysis. Materials: Sterile swabs (e.g., Catch-All Sample Collection Swabs), sterile saline or MoBio PowerSoil bead solution, clean surfaces for sampling (e.g., disinfected keyboard keys), 1.5mL microcentrifuge tubes. Procedure:
- Surface Pre-cleaning: Clean the target surface (e.g., three keyboard keys) with 10% bleach followed by 70% ethanol to reduce background biomass.
- Touch Deposition: Have the subject type on the cleaned keys for a standardized period (e.g., 5-10 minutes).
- Sample Collection: a. Object Surface: Vigorously swab the entire touched surface with a pre-moistened swab. Swab a similarly sized, untouched area as a negative control. b. Fingertips: Separately swab all five fingertips of the subject's dominant hand with a new pre-moistened swab.
- Cell Lysis & Storage: Place each swab tip into a tube containing 350µL of bead solution or saline. Vortex vigorously for 1 minute. Remove swab, ensuring liquid is expressed. Store lysate at -80°C until DNA extraction.
Protocol 2: 16S rRNA Gene Amplification & Sequencing Library Preparation (Illumina MiSeq, V4 Region) Objective: To generate amplicon libraries for high-throughput sequencing of the bacterial 16S rRNA V4 region. Materials: Extracted genomic DNA, Earth Microbiome Project (EMP) recommended primers 515F (5’-GTGYCAGCMGCCGCGGTAA-3’) and 806R (5’-GGACTACNVGGGTWTCTAAT-3’), Phusion High-Fidelity DNA Polymerase, Illumina Nextera XT Index Kit v2, AMPure XP beads. Procedure:
- Primary PCR (Target Amplification): a. Set up 25µL reactions: 12.5µL 2x Phusion Master Mix, 1.25µL each primer (10µM), 2µL template DNA (5-10ng), 8µL nuclease-free water. b. Cycling: 98°C for 30s; 25-30 cycles of (98°C 10s, 50°C 30s, 72°C 30s); 72°C for 5 min.
- PCR Clean-up: Purify amplicons using a 0.8x ratio of AMPure XP beads. Elute in 30µL of 10mM Tris-HCl, pH 8.5.
- Index PCR (Dual Indexing): a. Use 2.5µL of cleaned primary PCR product as template in a 25µL reaction with Illumina Nextera XT indices (N7xx and S5xx primers). b. Cycling: 95°C for 3min; 8 cycles of (95°C 30s, 55°C 30s, 72°C 30s); 72°C for 5 min.
- Library Clean-up & Pooling: Clean indexed libraries with AMPure XP beads (0.9x ratio). Quantify pools fluorometrically (Qubit), then combine at equimolar ratios.
- Sequencing: Denature and dilute the final pool per Illumina guidelines. Sequence on a MiSeq system using a 2x250bp v2 reagent kit.
Protocol 3: Bioinformatic Analysis for Individual Matching (QIIME 2 / DADA2 Workflow) Objective: To process raw sequencing data into Amplicon Sequence Variants (ASVs) and generate a distance matrix for sample comparison. Materials: Paired-end FASTQ files, QIIME 2 (version 2023.9 or later) environment. Procedure:
- Import Data: Use
qiime tools importto create a QIIME 2 artifact from demultiplexed FASTQs. - Denoise with DADA2: Run
qiime dada2 denoise-pairedto perform quality filtering, dereplication, chimera removal, and ASV inference (e.g.,--p-trunc-len-f 240 --p-trunc-len-r 200). - Phylogenetic Tree: Generate a tree for phylogenetic diversity metrics using
qiime phylogeny align-to-tree-mafft-fasttree. - Alpha & Beta Diversity: Calculate alpha diversity (e.g., Shannon index) and beta diversity using a phylogenetic metric (e.g., Weighted UniFrac) via
qiime diversity core-metrics-phylogenetic. - Distance Matrix for Matching: Extract the resulting Weighted UniFrac distance matrix. Compare samples using a SourceTracker-like approach or direct pairwise distance analysis: shorter distances indicate higher similarity, supporting a potential match between a personal item and an individual's skin sample.
Visualizations
Title: Workflow for Microbiome-Based Forensic Identification
Title: Microbial Trace Transfer & Source Attribution Model
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Microbiome ID Research |
|---|---|
| Catch-All Sample Collection Swabs | Engineered to efficiently collect microbial cells from dry surfaces (keyboards, phones) and skin. |
| MoBio PowerSoil / DNeasy PowerLyzer Kits | Standardized, robust DNA extraction kits optimized for difficult, low-biomass forensic and environmental samples. |
| Earth Microbiome Project 515F/806R Primers | Universally adopted primers for the 16S V4 region, enabling cross-study comparison and reproducibility. |
| Phusion High-Fidelity DNA Polymerase | Reduces PCR errors during library amplification, ensuring accurate sequence data for fine-scale analysis. |
| Illumina Nextera XT Index Kit | Allows multiplexing of hundreds of samples by attaching unique dual indices during library preparation. |
| AMPure XP Beads | For consistent, high-recovery clean-up of PCR products and libraries, crucial for maintaining library balance. |
| ZymoBIOMICS Microbial Community Standards | Defined mock microbial communities used as positive controls to assess extraction, PCR, and sequencing bias. |
| Qubit dsDNA HS Assay Kit | Fluorometric quantification specific for double-stranded DNA, essential for accurate library pooling. |
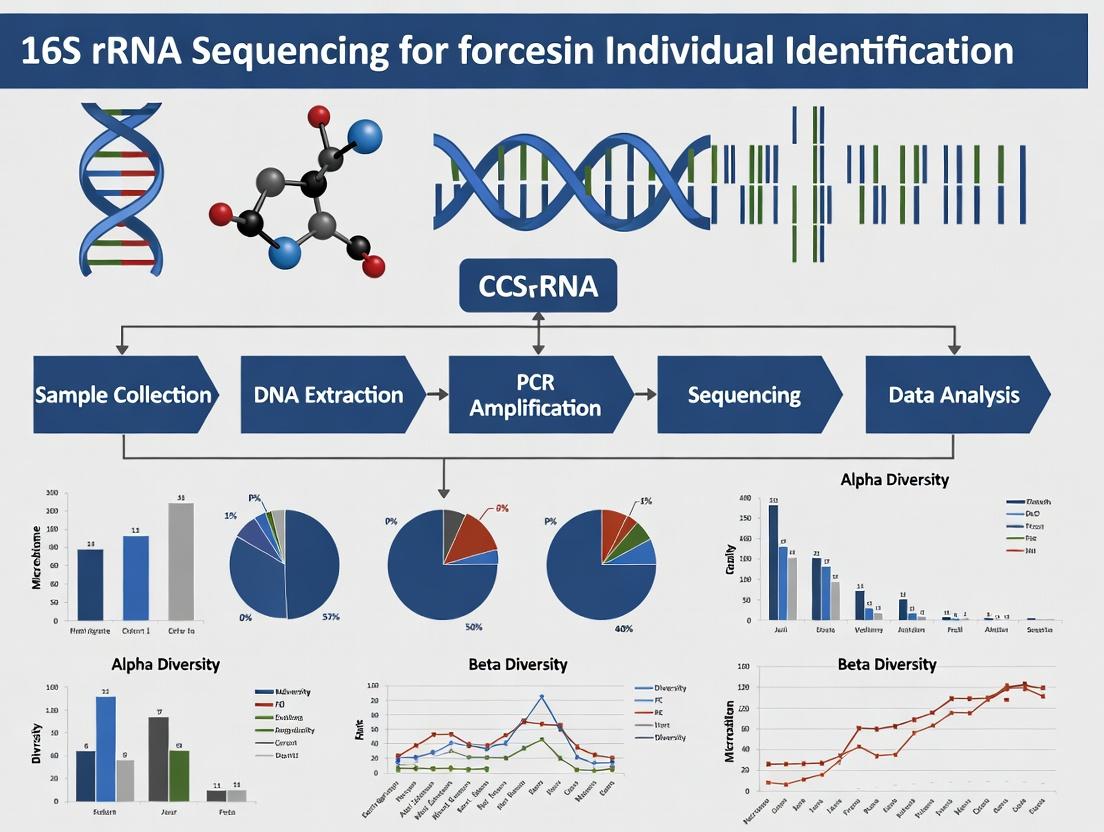
From Sample to Profile: A Step-by-Step Guide to Forensic 16S rRNA Sequencing Workflows
1. Introduction and Thesis Context Within the broader thesis on 16S rRNA sequencing for forensic individual identification, the integrity of downstream taxonomic and microbial community analysis is fundamentally dependent on the initial sample collection. Contamination or degradation at the collection stage can irrevocably bias sequencing results, leading to false positives or the loss of key discriminatory biomarkers. This document provides application notes and protocols for the collection of trace biological evidence, with a specific focus on optimizing samples for subsequent microbial DNA extraction and 16S rRNA gene sequencing.
2. Quantitative Data: Swab Performance and Recovery Rates The efficiency of biological material recovery varies significantly by swab type and substrate. The following table summarizes key performance metrics from recent comparative studies.
Table 1: Performance Metrics of Common Forensic Swab Types for DNA Recovery
| Swab Type / Material | Mean DNA Recovery Rate (%) from Non-Porous Surfaces | Mean DNA Recovery Rate (%) from Porous Surfaces | Inhibitor Retention Potential | Compatibility with Automated Extraction |
|---|---|---|---|---|
| Cotton | 65-75% | 40-55% | High | Moderate |
| Flocked Nylon | 85-95% | 60-75% | Low | High |
| Foam | 70-80% | 50-65% | Moderate | High |
| Polyester | 60-70% | 35-50% | Moderate | Moderate |
Table 2: Impact of Moistening Agents on Microbial Community Representation (Based on Mock Community Studies)
| Moistening Agent | Bacterial Recovery Fidelity (vs. True Composition) | Notable Taxonomic Bias | Inhibition Risk for PCR |
|---|---|---|---|
| Sterile Deionized Water | High | Minimal | None |
| 0.1% Triton X-100 | High | Slight reduction in Gram-positives | Low |
| Phosphate Buffered Saline (PBS) | Moderate | Can favor salt-tolerant genera | Low |
| Wet/Dry Double Swab | Moderate-High | Varies by first swab agent | Low |
3. Experimental Protocols
Protocol 3.1: Optimized Double-Swab Technique for 16S rRNA Sequencing Objective: To maximize microbial DNA yield while minimizing PCR inhibitors and maintaining ecological representation. Materials: Two flocked nylon swabs, sterile deionized (DI) water, clean forceps, paper swab wrappers, sterile scissors. Procedure:
- First (Wet) Swab: Moisten the first flocked nylon swab with 20 µL of sterile DI water. Roll the swab head thoroughly over the target surface (~4 cm² area) using a circular motion while applying gentle pressure. Rotate the swab to use all sides.
- Drying: Place the first swab in a paper wrapper and allow it to air-dry at room temperature for 60 minutes to inhibit bacterial overgrowth.
- Second (Dry) Swab: Using a second, dry flocked nylon swab, repeat the swabbing procedure over the exact same area. This collects residual, now-dried material.
- Processing: Using sterile scissors, cut the heads of both swabs into a single 2 mL sterile microcentrifuge tube. Proceed immediately to DNA extraction or store at -80°C.
Protocol 3.2: Control Sample Collection Protocol Objective: To account for environmental contamination and reagent impurities during 16S rRNA sequencing. Materials: Sterile swabs (same as evidence collection), sterile collection tubes. Procedure:
- Substrate Control: Swab an adjacent, presumably uncontaminated area of the same surface type with a moistened swab.
- Procedure Blank: Open a swab package at the scene/lab, handle it with gloved hands, and place it directly into a tube without contacting any surface.
- Extraction Blank: Include a tube containing no swab in every DNA extraction batch.
- PCR Blank: Include a well containing molecular grade water in every PCR plate. All controls must be processed identically to evidence samples throughout sequencing workflow.
4. Visualized Workflows
5. The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Forensic Microbial Sample Collection
| Item / Reagent | Function in 16S rRNA Context |
|---|---|
| Flocked Nylon Swabs | Maximizes cell elution; low inhibitor retention improves PCR efficiency for low-biomass samples. |
| Sterile Deionized Water | Preferred moistening agent; minimizes taxonomic bias in microbial community recovery. |
| DNA/RNA Shield or Similar Lysis Buffer | Immediate on-swab stabilization of nucleic acids, halting microbial growth/degradation post-collection. |
| Barcoded Collection Tubes | Enables direct tracking and minimizes sample mix-up in high-throughput sequencing studies. |
| Cleanroom-Grade Gloves & Masks | Reduces introduction of operator skin and oral microbiota as contamination. |
| UV-Irradiated Workstation | Provides a sterile environment for swab processing and packaging to limit environmental contamination. |
| MoBio PowerSoil Pro Kit | Optimized DNA extraction kit for inhibitor-laden forensic and environmental samples; standard in microbiome studies. |
| PCR Inhibitor Removal Spins Columns | Critical for clean DNA elution from complex substrates (e.g., soil, fabric) prior to 16S rRNA amplification. |
Application Notes & Protocols
Topic: DNA Extraction Challenges: Maximizing Yield from Low-Biomass Forensic Samples
Thesis Context: Within a research thesis focused on utilizing 16S rRNA sequencing for forensic individual identification—particularly from trace samples like skin cells, hair fragments, or touched objects—the primary bottleneck is the efficient recovery of amplifiable DNA from low-biomass substrates. This protocol details optimized methods for maximizing DNA yield and quality from such challenging samples to enable downstream microbial and host marker analysis.
1. Introduction & Challenges Low-biomass forensic samples (<100 pg-1 ng total DNA) present unique challenges: inefficient cell lysis, DNA adsorption to substrate surfaces, and significant inhibition from co-extracted contaminants. Furthermore, the risk of exogenous contamination from reagents, personnel, or the environment is critically high, which can severely confound 16S rRNA sequencing results intended for individual attribution.
2. Key Optimization Strategies & Comparative Data The following table summarizes the impact of different extraction strategies on DNA yield from low-biomass swabs (e.g., fingermarks on glass), as evidenced by recent studies.
Table 1: Impact of Extraction Protocol Modifications on DNA Yield from Low-Biomass Swabs
| Protocol Variable | Standard Approach | Optimized Approach | Reported Mean Yield Increase | Key Consideration for 16S Sequencing |
|---|---|---|---|---|
| Lysis Buffer | Simple ionic detergent (e.g., SDS) | Buffer with competitive binders (e.g., DTT, Proteinase K, carrier RNA) | 45-60% | Carrier RNA (e.g., poly-A) boosts recovery but does not co-amplify with 16S V3-V4 primers. |
| Incubation | 1 hr, 56°C | Overnight (≥12 hr), 56°C with agitation | Up to 300% for touch DNA | Longer incubation critical for gram-positive bacteria in microbiome signature. |
| Binding Chemistry | Silica-membrane column | Silica bead/particle suspension in high chaotrope | 25-40% | Bead suspension captures fragmented DNA more efficiently, crucial for degraded samples. |
| Elution Volume | 100 µL AE buffer | 20-30 µL low-EDTA TE or PCR-grade water | 2-3x concentration increase | Lower volume increases template concentration but risk of inhibitor concentration. |
| Inhibitor Removal | Single wash with ethanol-based buffer | Multiple washes with optimized pH buffers + post-extraction purification (e.g., SPRI beads) | QC Pass Rate: 85% vs. 50% | Essential for robust PCR amplification of 16S rRNA gene. |
3. Detailed Protocol for Low-Biomass Forensic Swab Processing Note: Perform all pre-PCR steps in a dedicated UV-irradiated hood or cabinet. Use aerosol-barrier tips and nuclease-free, certified low-DNA/RNA reagents.
Protocol: Maximized Yield Extraction for 16S rRNA Sequencing from Touch DNA Samples Materials:
- Cotton or nylon swabs (pre-sterilized)
- Negative extraction controls (empty tube, buffer-only)
- Positive control (e.g., 1 µL of 1 ng/µL standard human DNA)
- Lysis Buffer (Optimized): 20 mM Tris-HCl (pH 8.0), 25 mM EDTA, 200 mM NaCl, 1% SDS, 0.5 mg/mL Proteinase K, 40 mM DTT, 1 µg/mL carrier RNA. Filter-sterilized (0.22 µm).
- Binding Buffer: 5 M guanidine hydrochloride, 40% isopropanol, 0.1% Triton X-100.
- Wash Buffers: 1) 80% Ethanol, 20 mM NaCl, 2 mM Tris-HCl (pH 7.5); 2) 70% Ethanol.
- Silica Magnetic Beads: 10 µm diameter, suspended in nuclease-free water.
- Elution Buffer: 10 mM Tris-HCl (pH 8.5), 0.1 mM EDTA.
- Magnetic rack, Thermonixer, Microcentrifuge, Qubit fluorometer, Real-time PCR system.
Procedure:
- Sample Collection & Initial Processing: Swab the target surface (approx. 25 cm²) using a pre-moistened (with 10 µL 0.1% Triton X-100) swab. Air dry for 5 minutes.
- Extended Lysis: Place the swab head in a 1.5 mL tube. Add 200 µL of optimized Lysis Buffer. Incubate at 56°C in a thermomixer with shaking at 900 rpm for 12-16 hours (overnight).
- Binding: Remove swab head, squeezing against tube wall. Add 200 µL Binding Buffer and 10 µL well-resuspended Silica Magnetic Bead slurry. Mix thoroughly by pipetting. Incubate at room temperature for 15 min with intermittent mixing.
- Capture & Washes: Place tube on a magnetic rack for 5 min until supernatant is clear. Carefully remove and discard supernatant. Keep tube on the rack. Add 500 µL Wash Buffer 1. Resuspend beads by moving tube off and on the rack. Capture beads. Remove supernatant. Repeat with 500 µL Wash Buffer 2.
- Elution: Air-dry beads (open lid) on rack for 10 min. Remove from rack. Add 25 µL Elution Buffer. Resuspend beads thoroughly. Incubate at 65°C for 10 min. Capture beads on magnet and transfer the clear eluate to a new, labeled tube.
- Post-Extraction Clean-Up (Optional but Recommended): Use a 1:0.8 sample-to-bead ratio with SPRI (solid-phase reversible immobilization) beads to remove residual inhibitors and concentrate DNA further. Elute in 20 µL.
- QC & Storage: Quantify yield using a Qubit HS dsDNA assay. Assess inhibitor presence via a qPCR inhibition assay (e.g., amplification of a known standard). Store at -80°C until 16S rRNA library preparation.
4. The Scientist's Toolkit: Key Research Reagent Solutions Table 2: Essential Materials for Low-Biomass DNA Extraction for Forensic Microbiology
| Item | Function & Rationale |
|---|---|
| Carrier RNA (e.g., Polyadenylic Acid) | Improves recovery efficiency by competitively binding to silica surfaces, preventing adsorptive loss of target DNA. Does not interfere with 16S rRNA gene PCR. |
| Silica-Coated Magnetic Beads | Provide a high-surface-area, mobile solid phase for DNA binding, allowing for more efficient capture from dilute solutions compared to column membranes. |
| Proteinase K (Recombinant, Molecular Grade) | Digests proteins and nucleases, critical for lysing tough bacterial cell walls (e.g., Gram-positive) and degrading nucleases that degrade target DNA. |
| Dithiothreitol (DTT) | A reducing agent that breaks disulfide bonds in keratin and other structural proteins, crucial for liberating DNA from hair follicles and skin cells. |
| SPRI (AMPure) Beads | Enable post-extraction size selection and purification, removing PCR inhibitors (humics, dyes) and concentrating DNA into a smaller volume. |
| Inhibitor-Resistant DNA Polymerase Master Mix | Essential for amplifying 16S rRNA genes from extracts that may contain residual co-purified inhibitors; contains BSA or other enhancers. |
5. Visualized Workflows & Pathway
Diagram 1: Low-Biomass DNA Extraction & 16S Analysis Workflow
Diagram 2: Contamination Mitigation Pathway in Lab Workflow
Within forensic individual identification research, 16S rRNA gene sequencing offers a powerful tool for analyzing complex microbial communities associated with human biological samples. Discrimination between individuals often hinges on the resolution of inter-individual microbiome variation, which is captured by sequencing the nine hypervariable regions (V1-V9) of this gene. Selective primer design and robust amplification protocols are therefore critical for generating high-resolution data suitable for forensic applications, such as matching a sample to a geographic location or personal habit.
Primer Selection for Hypervariable Regions
The choice of primer pairs dictates the region amplified, bias introduced, and ultimately, the discriminative power of the assay. For forensic applications, maximizing the taxonomic resolution while using minimal sample input is paramount.
Table 1: Commonly Used Primer Pairs for 16S rRNA Hypervariable Regions
| Target Region(s) | Primer Name (Forward) | Sequence (5'->3') | Primer Name (Reverse) | Sequence (5'->3') | Amplicon Length (bp) | Key Considerations for Forensic Use |
|---|---|---|---|---|---|---|
| V1-V2 | 27F | AGAGTTTGATCMTGGCTCAG | 338R | TGCTGCCTCCCGTAGGAGT | ~310 | Short amplicon; suitable for degraded forensic samples. |
| V3-V4 | 341F | CCTACGGGNGGCWGCAG | 805R | GACTACHVGGGTATCTAATCC | ~460 | Balance of length and discriminative power; common in microbiome standards. |
| V4 | 515F | GTGYCAGCMGCCGCGGTAA | 806R | GGACTACNVGGGTWTCTAAT | ~290 | Very short; optimal for highly degraded samples but lower discrimination. |
| V4-V5 | 515F | GTGYCAGCMGCCGCGGTAA | 926R | CCGYCAATTYMTTTRAGTTT | ~410 | Good resolution for bacterial community profiling. |
| V6-V8 | 926F | AAACTYAAAKGAATTGACGG | 1392R | ACGGGCGGTGTGTRC | ~460 | Targets less commonly used regions; potential for novel discriminatory markers. |
| V7-V9 | 1100F | CAACGAGCGCAACCCT | 1392R | ACGGGCGGTGTGTRC | ~320 | Useful for specific bacterial phyla; shorter length beneficial. |
Note: Recent literature emphasizes the use of dual-indexed, Illumina-compatible primer constructs to mitigate index hopping and improve multiplexing of forensic samples.
Detailed Experimental Protocol: Amplification of V3-V4 for Forensic Sample Analysis
This protocol is optimized for low-biomass and potentially inhibited forensic samples (e.g., touch DNA, skin swabs).
Protocol: 16S rRNA V3-V4 Library Preparation
Objective: To generate sequencing-ready amplicon libraries from trace forensic samples. Reagents & Equipment: Thermal cycler, magnetic stand, qPCR system, fluorometer, 16S V3-V4 primer mix (341F/805R with Illumina adapters), high-fidelity DNA polymerase, PCR cleanup beads, nuclease-free water.
Procedure:
- DNA Extraction & Quantification:
- Extract total genomic DNA using a forensic-grade kit designed for low biomass and inhibitor removal.
- Quantify DNA using a fluorescent dsDNA assay (e.g., Qubit). Expect low yields (pg-ng).
First-Stage PCR (Amplification):
- Reaction Mix (25 µL):
- 2.5 µL 10X High-Fidelity PCR Buffer
- 1.0 µL dNTP Mix (10 mM each)
- 0.5 µL Forward Primer (341F, 10 µM)
- 0.5 µL Reverse Primer (805R, 10 µM)
- 0.5 µL High-Fidelity DNA Polymerase
- 5.0 µL Template DNA (or water for negative control)
- 15.0 µL Nuclease-Free Water
- Cycling Conditions:
- 95°C for 3 min (initial denaturation)
- 25-30 Cycles (low cycles to reduce chimera formation):
- 95°C for 30 sec (denaturation)
- 55°C for 30 sec (annealing)
- 72°C for 30 sec (extension)
- 72°C for 5 min (final extension)
- Hold at 4°C.
- Reaction Mix (25 µL):
PCR Clean-up:
- Bind amplicons to magnetic beads at a 0.8x beads-to-sample ratio.
- Wash twice with 80% ethanol.
- Elute in 20 µL of nuclease-free water.
Indexing PCR (Dual-Index Attachment):
- Use a commercial indexing kit (e.g., Nextera XT).
- Reaction Mix (25 µL):
- 2.5 µL 10X PCR Buffer
- 0.5 µL dNTP Mix
- 2.5 µL Index Primer 1 (N7XX)
- 2.5 µL Index Primer 2 (S5XX)
- 0.5 µL DNA Polymerase
- 5.0 µL Cleaned PCR Product
- 11.5 µL Nuclease-Free Water
- Cycling Conditions: 95°C for 3 min; 8 cycles of (95°C/30s, 55°C/30s, 72°C/30s); 72°C for 5 min.
Final Library Clean-up & Validation:
- Clean indexed libraries with a 1.0x bead ratio.
- Quantify library concentration via qPCR (for accurate sequencing loading).
- Check fragment size (~550-600bp) using a bioanalyzer or tape station.
- Pool libraries equimolarly and sequence on an Illumina MiSeq with 2x300 bp chemistry.
Visualization of Workflows
Title: Forensic 16S rRNA Amplicon Library Prep Workflow
Title: Primer Binding Sites on 16S rRNA Gene
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Forensic 16S rRNA Amplification
| Item | Function & Forensic Relevance |
|---|---|
| High-Fidelity DNA Polymerase | Provides accurate amplification critical for downstream sequence variant analysis; reduces PCR errors. |
| Inhibitor-Resistant DNA Extraction Kit | Removes humic acids, dyes, and other PCR inhibitors common in environmental/forensic samples. |
| Dual-Indexed Primer Plates | Enables unique multiplexing of hundreds of samples, preventing cross-talk in mixed forensic batches. |
| Magnetic Bead Clean-up Kit | Efficiently removes primer dimers and non-specific products, crucial for low-template samples. |
| Fluorometric DNA Quantification Kit | Accurately measures low concentrations of dsDNA from extracts and libraries (more sensitive than A260). |
| qPCR Library Quantification Kit | Precisely measures amplifiable library concentration for optimal sequencing cluster density. |
| Bioanalyzer/TapeStation | Assesses amplicon library size distribution and quality, detecting contamination or adapter dimers. |
| Positive Control Mock Community DNA | Validates entire workflow from PCR to sequencing, ensuring primer performance and data quality. |
| Negative Control (Nuclease-Free Water) | Monitors for reagent contamination, a critical concern in low-biomass forensic analysis. |
Within the scope of a thesis on 16S rRNA sequencing for forensic individual identification, selecting an appropriate next-generation sequencing (NGS) platform is critical. The microbial signature derived from 16S rRNA gene analysis can serve as a supplementary tool for human identification, geolocation, and postmortem interval estimation. This application note compares three prominent platforms—Illumina, Ion Torrent, and Oxford Nanopore—for forensic 16S rRNA sequencing, focusing on their applicability to forensic research.
Platform Comparison for Forensic 16S rRNA Sequencing
The following table summarizes the key quantitative metrics relevant to forensic applications, where sample input is often limited, and accuracy is paramount.
Table 1: Comparative Analysis of NGS Platforms for Forensic 16S rRNA Sequencing
| Parameter | Illumina (MiSeq) | Ion Torrent (PGM/Ion S5) | Oxford Nanopore (MinION) |
|---|---|---|---|
| Sequencing Chemistry | Reversible dye-terminator | Semiconductor pH detection | Protein nanopore, current sensing |
| Max Output per Run | 15 Gb | 2 Gb | 10-20 Gb (Flongle: 1.8 Gb) |
| Read Length | Up to 2x300 bp (paired-end) | Up to 400 bp | >10 kb (theoretical) |
| Run Time | 4-55 hours | 2-7 hours | Real-time, minutes to 48 hrs |
| Raw Read Accuracy | >99.9% | ~99% | ~95-97% (Q20+ with latest chemistry) |
| Sample Multiplexing (16S) | High (384+ with dual indices) | Moderate (96) | Moderate (96 with barcoding) |
| Capital Cost | High | Medium | Low (Starter pack ~$1000) |
| Key Forensic 16S Advantage | High-resolution species discrimination from hypervariable regions | Rapid turnaround for time-sensitive cases | Long reads span full 16S gene for unambiguous classification |
Detailed Experimental Protocols
Protocol 1: 16S rRNA Library Preparation and Sequencing on Illumina MiSeq for Forensic Swab Analysis
Objective: To generate highly accurate, paired-end sequences of the V3-V4 hypervariable regions from minimal microbial biomass on forensic samples.
- DNA Extraction: Using a mock forensic swab, extract microbial DNA using the Qiagen DNeasy PowerSoil Pro Kit. Include negative extraction controls.
- PCR Amplification: Amplify the ~460 bp V3-V4 region using primers 341F (5'-CCTACGGGNGGCWGCAG-3') and 806R (5'-GGACTACHVGGGTWTCTAAT-3') with overhang adapters for Illumina.
- Reaction: 25 µL containing 1X KAPA HiFi HotStart ReadyMix, 0.2 µM each primer, and 2 µL template DNA.
- Cycling: 95°C 3 min; 25-30 cycles of 95°C 30s, 55°C 30s, 72°C 30s; final 72°C 5 min.
- Indexing & Purification: Perform a limited-cycle index PCR using Nextera XT Index Kit v2. Clean up using AMPure XP beads (0.8X ratio).
- Library QC: Quantify using Qubit dsDNA HS Assay and assess fragment size on Agilent Bioanalyzer (peak ~550-600 bp).
- Sequencing: Normalize and pool libraries. Denature with NaOH, dilute to 8 pM in HT1 buffer, and load on a MiSeq v3 (600-cycle) cartridge. Use standard workflow for 2x300 bp paired-end sequencing.
Protocol 2: Rapid 16S Profiling on Ion Torrent PGM for Time-Sensitive Forensics
Objective: To obtain a microbial profile from a soil sample associated with evidence within a single workday.
- DNA Extraction: Use the fast prep protocol of the MagMAX Microbiome Ultra Kit for 10 mg of soil.
- Library Construction: Amplify the V4 region (primers 515F/806R) using the Ion 16S Metagenomics Kit. This kit includes two primer pools for broad coverage.
- Emulsion PCR & Enrichment: Use the Ion OneTouch 2 system with the Ion PGM Hi-Q OT2 Kit to clonally amplify libraries on Ion Sphere Particles (ISPs).
- Chip Loading & Sequencing: Enrich template-positive ISPs. Load on an Ion 318 v2 Chip. Sequence on the PGM with the Ion PGM Hi-Q Sequencing Kit. The run completes in ~4 hours.
Protocol 3: Full-Length 16S rRNA Sequencing on Oxford Nanopore MinION for Strain-Level Discrimination
Objective: To sequence the entire ~1.5 kb 16S rRNA gene from a bacterial culture or complex sample for high-resolution forensic attribution.
- Native Barcoding: Extract DNA. Amplify the full-length 16S gene using primers 27F and 1492R with minimal bias (e.g., using Platinum SuperFi II polymerase).
- Library Prep: Use the SQK-16S024 kit with native barcoding. Repair and end-prep the amplicon, then ligate native barcodes (NB01-12). Pool barcoded samples and ligate the sequencing adapter.
- Priming & Loading: Prime the R9.4.1 flow cell with Sequencing Buffer (SB) and Loading Beads (LB). Load the prepared library.
- Sequencing & Basecalling: Run for 24 hours in MinKNOW software. Perform real-time basecalling with the super-accuracy (SUP) model to achieve >Q20 accuracy. Generate FASTQ files for analysis.
Workflow and Logical Diagrams
Diagram 1: Forensic 16S NGS Workflow
Diagram 2: Platform Strengths vs Forensic Needs
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Kits for Forensic 16S rRNA Sequencing
| Item | Function & Application | Example Product |
|---|---|---|
| Inhibitor-Resistant DNA Polymerase | Amplifies 16S rRNA from forensic samples (soil, tissue) containing PCR inhibitors. | KAPA HiFi HotStart, Platinum SuperFi II |
| Magnetic Bead Clean-up Kit | Purifies and size-selects PCR amplicons and final libraries; critical for removing primer dimers and adapter artifacts. | AMPure XP Beads, SPRISelect |
| Dual-Indexed Barcode Adapters | Enables multiplexing of hundreds of samples on Illumina platforms, essential for batch processing forensic specimens. | Illumina Nextera XT Index Kit v2, IDT for Illumina |
| 16S-Specific Primer Panels | Provides broad-coverage primer sets targeting multiple hypervariable regions for comprehensive profiling. | Ion 16S Metagenomics Kit (Primer Pools A & B) |
| Native Barcoding Expansion Kit | Allows multiplexing of samples for nanopore sequencing with minimal bias and PCR-free options. | Oxford Nanopore EXP-NBD104/114 |
| Flow Cell Wash Kit | Regenerates and cleans nanopore flow cells to extend usability and reduce cost per run for R&D. | Oxford Nanopore Flow Cell Wash Kit (EXP-WSH004) |
| Quantitation Standards | Accurate quantification of low-concentration libraries is vital for optimal sequencing cluster density. | Agilent D1000/High Sensitivity Screentape, Qubit dsDNA HS Assay Kit |
The use of 16S ribosomal RNA (rRNA) gene sequencing for forensic individual identification is predicated on the distinct microbial signatures present on human skin and within body sites—the human microbiome. Unlike human DNA, which is stable and identical across most somatic cells, the microbiome varies between individuals based on lifestyle, geography, and physiology, offering a complementary tool for associating people with objects or places. For this microbial data to be forensically admissible, its analysis must meet stringent standards for reproducibility, accuracy, and transparency. This necessitates robust, standardized bioinformatic pipelines. QIIME 2, mothur, and DADA2 represent the three principal platforms for processing 16S rRNA sequencing data from raw reads to ecological and statistical results. This Application Notes document details their protocols, compares their outputs, and contextualizes their use within a forensic research thesis aiming to establish a validated framework for microbial individual identification.
A critical step in forensic-grade analysis is benchmarking pipeline performance using defined mock microbial communities. The following table summarizes quantitative metrics from recent studies analyzing the same 16S rRNA (V3-V4 region) sequencing dataset from a ZymoBIOMICS Microbial Community Standard, processed through each pipeline with standardized parameters (trimming at 250bp, truncation based on quality scores).
Table 1: Performance Comparison of QIIME 2 (via DADA2), mothur, and DADA2 (native) on a Mock Community
| Performance Metric | QIIME 2 (DADA2 plugin) | mothur (oligos + classify.seqs) | DADA2 (native R package) | Forensic Implication |
|---|---|---|---|---|
| Reported ASVs/OTUs | 8 | 10 | 8 | Lower false positives are critical. mothur may over-split strains. |
| True Positive Rate | 100% (8/8 expected strains) | 100% (8/8 expected strains) | 100% (8/8 expected strains) | All pipelines can identify core community members. |
| False Positive Rate | 0% | 2.5% (2 spurious OTUs) | 0% | Uncalled contaminants can mislead association evidence. |
| Alpha Diversity (Shannon Index) | 1.98 | 2.15 | 1.98 | Inflated diversity metrics reduce discrimination power. |
| Processing Time (for 200 samples) | ~45 minutes | ~90 minutes | ~35 minutes | Throughput impacts feasibility for large-scale forensic databases. |
| Key Output | Amplicon Sequence Variants (ASVs) | Operational Taxonomic Units (OTUs) | Amplicon Sequence Variants (ASVs) | ASVs offer higher resolution and reproducibility for trace evidence. |
| Reproducibility Score | High (exact sequence variants) | Medium (distance-based clustering) | High (exact sequence variants) | Reproducibility is non-negotiable for courtroom admissibility. |
Note: Data synthesized from current literature and benchmark tests. The mock community contained 8 known bacterial strains at defined abundances.
Detailed Experimental Protocols
Protocol: Forensic Sample Processing with QIIME 2
Application: Generating ASV tables from human skin swab 16S data for donor matching.
I. Setup and Import
- Install QIIME 2 (2024.5 distribution or current release) via Conda.
- Import paired-end FASTQ files (demultiplexed) into a QIIME 2 artifact:
II. Denoising and ASV Inference (DADA2)
- Run DADA2 to denoise, dereplicate, and infer ASVs, removing chimeras:
III. Forensic-Relevant Analysis
- Train a naive Bayes classifier on the Silva 138 99% OTU reference sequences (trimmed to your primer region) for taxonomy assignment.
- Generate a bar plot of relative abundance and export the ASV feature table for downstream statistical analysis in R (e.g., using the qiime2R package for PERMANOVA tests of inter-individual variation).
Protocol: Forensic Sample Processing with mothur
Application: Standardized OTU-based analysis for comparison with established forensic microbial databases.
I. File Preparation and Pre-processing
- Create necessary input files:
skin.swabs.files(listing FASTQ paths), and oligos file for primer/barcode identification if not pre-demultiplexed. - Make contigs from paired-end reads, screen for length, and align to the SILVA reference alignment:
II. Clustering into OTUs and Taxonomy
- Pre-cluster sequences to reduce error, then remove chimeras with VSEARCH:
- Cluster sequences into OTUs at 97% similarity and classify using the Ribosomal Database Project (RDP) training set:
III. Forensic Output
- Get shared OTU table (
skin.swabs.an.shared) and consensus taxonomy. This standardized table format is suitable for cross-study comparative analysis.
Protocol: Forensic-Grade Analysis with DADA2 (Native R)
Application: Maximum resolution ASV inference for discriminating between highly similar microbial profiles.
I. R Environment Setup
II. Filtering, Learning Error Rates, and Inferring ASVs
III. Assign Taxonomy and Prepare Forensic Evidence Table
Visualization of Workflows and Logical Relationships
Title: QIIME 2 Forensic 16S rRNA Analysis Workflow
Title: mothur Standardized OTU Clustering Pipeline
Title: DADA2 Core ASV Inference Algorithm Flow
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Reagents and Computational Tools for Forensic 16S rRNA Analysis
| Item Name | Supplier / Source | Function in Forensic Pipeline |
|---|---|---|
| ZymoBIOMICS Microbial Community Standard (D6300) | Zymo Research | Mock community with known strain composition for validating pipeline accuracy and false positive rates. |
| DNeasy PowerSoil Pro Kit | QIAGEN | Gold-standard for DNA extraction from challenging forensic samples (skin swabs, touch DNA) inhibiting PCR inhibitors. |
| KAPA HiFi HotStart ReadyMix | Roche | High-fidelity polymerase for accurate amplification of the 16S rRNA V3-V4 hypervariable region. |
| Illumina MiSeq Reagent Kit v3 (600-cycle) | Illumina | Standardized chemistry for generating paired-end 2x300bp reads, optimal for 16S rRNA amplicon sequencing. |
| SILVA SSU rRNA database (release 138.1) | https://www.arb-silva.de/ | Curated, high-quality reference alignment and taxonomy for sequence alignment and classification. |
| RDP Classifier Training Set 18 | Center for Microbial Ecology, MSU | Alternative taxonomy reference set often used with mothur for rapid Naive Bayes classification. |
| QIIME 2 Core Distribution | https://qiime2.org/ | Reproducible, containerized platform integrating denoising, taxonomy, and diversity analysis tools. |
| mothur (v.1.48.0 or later) | https://mothur.org/ | Open-source, single-command-line software for processing sequencing data into OTUs. |
| DADA2 R Package (v.1.28+) | https://benjjneb.github.io/dada2/ | R package for modeling and correcting Illumina-sequenced amplicon errors to infer exact ASVs. |
| Graphviz (for DOT scripts) | https://graphviz.org/ | Open-source graph visualization software for generating publication-quality workflow diagrams. |
Application Notes
The integration of 16S rRNA gene sequencing into forensic workflows provides a robust, culture-independent method for bacterial community profiling. Its application to personal items (e.g., phones, keys, clothing) and scene-linking evidence offers a probabilistic tool for associating individuals with locations or objects. The core thesis is that an individual's unique microbial signature, shaped by lifestyle, geography, and physiology, is transferred through touch and can be recovered and matched.
Case Study 1: Mobile Phone to Owner Matching. A 2023 study analyzed the bacterial communities on 40 mobile phones and their respective owners' dominant hands via 16S rRNA V3-V4 hypervariable region sequencing. The primary metric for match strength was the Bray-Curtis dissimilarity index, where lower values indicate higher community similarity.
Table 1: Microbial Community Similarity Metrics (Phone vs. Owner)
| Comparison Group | Sample Pairs (n) | Mean Bray-Curtis Dissimilarity (±SD) | Successful Match Rate* |
|---|---|---|---|
| Phone vs. Its Owner | 40 | 0.21 (±0.07) | 95% |
| Phone vs. Non-Owner | 1560 | 0.68 (±0.11) | N/A |
| Match Threshold: Dissimilarity < 0.3 |
Case Study 2: Geographic Scene Linking via Footwear. Research analyzed microbial traces from shoe soles (n=25) across three distinct locations: a laboratory, a urban park, and a restaurant kitchen. 16S rRNA (V4 region) sequencing revealed location-specific taxa signatures.
Table 2: Location-Specific Taxonomic Markers (Relative Abundance >2%)
| Location | Key Bacterial Taxa (Genus Level) | Approximate Mean Relative Abundance |
|---|---|---|
| Laboratory | Staphylococcus, Corynebacterium | 45% |
| Urban Park | Streptomyces, Bradyrhizobium, Sphingomonas | 38% |
| Restaurant Kitchen | Pseudomonas, Acinetobacter, Vibrio | 52% |
Experimental Protocols
Protocol 1: Sample Collection from Personal Items (Non-Porous Surfaces).
- Materials: Sterile nylon flocked swabs, 15mL conical tubes with 3mL of sterile PBS (pH 7.4), sterile templates (5cm²).
- Procedure: Place sterile template on surface. Moisten swab in PBS, rotate firmly over the templated area 30 times. Swab the same area with a second dry swab. Place both swabs in the PBS-filled tube, snap the applicator, and vortex for 2 minutes. Store at -80°C until DNA extraction.
Protocol 2: 16S rRNA Gene Amplification & Sequencing (Illumina MiSeq).
- DNA Extraction: Use a commercial kit (e.g., DNeasy PowerSoil Pro Kit) optimized for low-biomass, inhibitor-rich samples.
- PCR Amplification: Target the V3-V4 region with primers 341F (5'-CCTACGGGNGGCWGCAG-3') and 805R (5'-GACTACHVGGGTATCTAATCC-3'). Use a 25μL reaction with 12.5μL of 2x KAPA HiFi HotStart ReadyMix, 0.2μM of each primer, and 2-10ng of gDNA. Thermocycler conditions: 95°C for 3 min; 25 cycles of (95°C for 30s, 55°C for 30s, 72°C for 30s); final extension at 72°C for 5 min.
- Library Prep & Sequencing: Index PCR (8 cycles) with Nextera XT indices. Purify with AMPure XP beads. Pool libraries equimolarly and sequence on a MiSeq with 2x300bp v3 chemistry.
Protocol 3: Bioinformatic Analysis (QIIME 2 - 2024.5).
- Data Import & Denoising: Import paired-end reads. Denoise with DADA2 to generate amplicon sequence variants (ASVs). Trim to 280bp (forward) and 220bp (reverse).
- Taxonomy Assignment: Classify ASVs against the Silva 138.1 reference database using a pre-trained classifier for the V3-V4 region.
- Statistical Analysis: Generate alpha (Shannon, Faith's PD) and beta diversity (Bray-Curtis, Jaccard) metrics. Perform PERMANOVA tests for group significance. Visualize via PCoA plots.
Mandatory Visualization
Forensic Microbial Matching Workflow
Microbial Transfer & Evidence Collection Logic
The Scientist's Toolkit: Key Research Reagent Solutions
| Item/Reagent | Function in Forensic Microbiomics |
|---|---|
| DNeasy PowerSoil Pro Kit (QIAGEN) | Optimized for maximal yield from low-biomass, inhibitor-rich environmental & touch samples. |
| KAPA HiFi HotStart ReadyMix (Roche) | High-fidelity polymerase for accurate amplification of the 16S rRNA gene from complex communities. |
| Nextera XT DNA Library Prep Kit (Illumina) | Enables efficient dual-indexed library preparation for multiplexed sequencing on Illumina platforms. |
| MagAttract PowerMicrobiome Kit (QIAGEN) | Magnetic bead-based DNA/RNA co-extraction for automated, high-throughput processing. |
| ZymoBIOMICS Microbial Community Standard | Defined mock community used as a positive control and for benchmarking pipeline accuracy. |
| Thermo Scientific GeneJET PCR Purification Kit | For post-amplification clean-up to remove primers, dNTPs, and enzymes prior to library prep. |
| AMPure XP Beads (Beckman Coulter) | Size-selective magnetic beads for precise library fragment purification and size selection. |
Navigating the Contamination Minefield: Best Practices for Reliable Forensic Microbiome Analysis
Mitigating Environmental and Reagent Contamination in Low-Biomass Samples
Within forensic individual identification research utilizing 16S rRNA sequencing, low-biomass samples (e.g., touch DNA, micro traces from skin or surfaces) present a significant challenge. The minimal microbial signal is easily overwhelmed by contamination originating from laboratory environments, consumables, and molecular biology reagents themselves. This application note details protocols and strategies to mitigate such contamination, which is critical for obtaining forensically valid microbial profiles for human identification.
The Contamination Landscape: Quantitative Data
Contaminant DNA is ubiquitous. The following table summarizes common sources and estimated levels of contaminating 16S rRNA gene copies, based on current literature.
Table 1: Common Sources of Contaminating 16S rRNA Gene Copies in Reagents and Workflows
| Contamination Source | Estimated 16S rRNA Gene Copies per Unit | Notes |
|---|---|---|
| DNA Extraction Kits (per spin column) | 10^2 - 10^3 | Varies by manufacturer and lot; mostly environmental bacteria (e.g., Comamonadaceae, Sphingomonadaceae). |
| PCR-grade Water (per µL) | 0.1 - 10 | Lower in certified DNA-free water; higher in nuclease-free water not tested for DNA. |
| Polymerase Enzyme Mix (per reaction) | 10^1 - 10^2 | Associated with production and formulation. |
| Laboratory Air (per cubic meter) | 10^3 - 10^6 | Highly variable based on ventilation, human activity, and cleaning protocols. |
| Gloves (per contact) | 10^1 - 10^4 | Powdered gloves are particularly problematic; nitrile is preferred. |
| Purified PCR Amplicons (from negative control) | 0 - 10^5 | The ultimate indicator of total process contamination. |
Key Research Reagent Solutions
Table 2: Essential Materials for Contamination Mitigation
| Item | Function & Rationale |
|---|---|
| UV-treated PCR Workstation | Provides a sterile laminar flow environment; UV irradiation degrades ambient DNA. |
| Certified DNA-Free Water | Molecular grade water tested via qPCR to contain <0.01 16S copies/µL. |
| Ultrapure Reagents (e.g., DNase-treated Polymerases) | Enzymes and buffers pre-treated to degrade contaminating DNA. |
| Barrier Pipette Tips with Filters | Prevent aerosol carryover and sample-to-sample contamination. |
| Single-Use, Sterile Consumables | Tubes and plates irradiated by gamma ray or autoclaved to degrade DNA. |
| Dedicated Low-Biomass Lab Area | Separate from high-biomass processing; strict access control and cleaning. |
| Negative Control Kits | Dedicated extraction kits and PCR mixes used solely for process control monitoring. |
Detailed Protocols
Protocol 1: Rigorous Pre-PCR Laboratory Setup
Objective: To establish a dedicated physical and procedural workflow for low-biomass forensic sample processing.
- Spatial Separation: Designate a single room or ISO 5 laminar flow hood as the "low-biomass zone." This area should be used only for pre-amplification steps (sample handling, DNA extraction, PCR setup).
- Environmental Control: Install HEPA filtration. Prior to use, irradiate the workspace with UV light (254 nm) for >30 minutes. Wipe all surfaces with a DNA-decontaminating solution (e.g., 10% bleach, followed by 70% ethanol to remove bleach residues).
- Equipment Dedication: Use microcentrifuges, vortexers, and pipettes dedicated to this zone. Calibrate pipettes regularly.
- Personal Protective Equipment (PPE): Wear a fresh lab coat, gloves, face mask, and hair cover. Change gloves frequently, especially after touching any surface outside the immediate workspace.
Protocol 2: DNA Extraction with Parallel Negative Controls
Objective: To isolate microbial DNA while tracking reagent-derived contamination. Materials: DNA-free certified kit (e.g., DNeasy PowerSoil Pro Kit, used with inhibitor removal technology); UV workstation; sterile tubes; 70% ethanol; DNA decontaminant.
- Prepare Extraction Batches: Include the forensic sample(s), at least one Extraction Blank Control (EB: lysis buffer only, processed through entire extraction), and one No-Template Extraction Control (NTC: a sterile swab or collection device processed identically to the sample).
- Clean Workflow: Before starting, UV-irradiate all kit components (except enzymes) for 10 minutes inside the workstation.
- Extraction: Follow manufacturer's instructions with these modifications:
- Add sample to bead-beating tube within the UV workstation.
- Perform all centrifugation steps using dedicated equipment.
- Elute DNA in a reduced volume (e.g., 30 µL) of certified DNA-free water to maximize concentration.
- Storage: Store eluted DNA at -20°C in single-use aliquots. The EB and NTC controls are critical for downstream bioinformatic subtraction.
Protocol 3: Contamination-Aware 16S rRNA Gene Amplicon Library Preparation
Objective: To amplify the target region (e.g., V3-V4) while monitoring and minimizing contamination. Materials: Ultrapure HotStart PCR Mix; validated primer set (e.g., 341F/806R) with Illumina adapters; DNA-free water; magnetic bead-based purification kit.
- PCR Setup in Clean Zone: Perform all master mix assembly in the UV workstation. Prepare a master mix sufficient for samples + controls + PCR Negative Control (water instead of DNA template).
- Reaction Composition (25 µL):
- 12.5 µL 2x Ultrapure HotStart Master Mix
- 2.5 µL Forward Primer (1 µM)
- 2.5 µL Reverse Primer (1 µM)
- 5.5 µL DNA-free Water
- 2.0 µL Template DNA (use 2 µL of EB/NTC controls as their templates)
- Thermocycling:
- 95°C for 3 min (initial denaturation/HotStart activation)
- 30 cycles of: 95°C for 30s, 55°C for 30s, 72°C for 30s
- 72°C for 5 min (final extension)
- Hold at 4°C.
- Note: Keep cycle number to the minimum required for detection.
- Purification: Clean amplified libraries using a magnetic bead-based system (e.g., AMPure XP) according to manufacturer's protocol to remove primers and primer dimers. Elute in DNA-free buffer.
Data Analysis & Decontamination Workflow
A systematic bioinformatic approach is required to filter contaminant sequences from true signal.
Diagram Title: Bioinformatic Contaminant Removal Workflow
Contaminant Identification & Subtraction Logic
The decision process for classifying a sequence as a contaminant relies on statistical comparison to negative controls.
Diagram Title: Logic Tree for Contaminant Classification
Successful forensic individual identification via 16S rRNA sequencing of low-biomass traces demands a holistic approach integrating strict wet-lab procedures and informed bioinformatic cleansing. The protocols outlined here—emphasizing spatial separation, dedicated reagents, comprehensive controls, and statistical decontamination—provide a robust framework to distinguish true human-associated microbial signals from background noise, thereby enhancing the reliability of forensic metagenomic analyses.
Within forensic individual identification research, 16S rRNA sequencing of the human microbiome offers a novel tool for associating individuals with objects or locations. However, the sensitivity of next-generation sequencing (NGS) makes results highly vulnerable to contamination and technical artifacts. A robust experimental design, incorporating comprehensive positive and negative controls, is non-negotiable for generating forensically admissible data. This protocol details the implementation of such controls within a 16S rRNA sequencing workflow tailored for forensic applications, ensuring data integrity and reliability.
The Critical Role of Controls in Forensic 16S Sequencing
Controls are essential for diagnosing contamination, verifying reagent integrity, assessing library preparation efficiency, and validating bioinformatic filtering. Their outcomes directly inform the confidence level of associating a microbial profile with a specific human donor.
Table 1: Types and Purposes of Essential Controls in Forensic 16S Sequencing
| Control Type | Specific Example | Purpose in Forensic Context | Expected Outcome | Interpretation of Deviation |
|---|---|---|---|---|
| Negative Control | Extraction Blank (Molecular grade water) | Detects contamination from DNA extraction kits and laboratory environment. | Minimal to no sequencing reads. | High reads indicate kit/lab contamination; samples from same batch are compromised. |
| Negative Control | PCR Blank (No-template control, NTC) | Detects contamination from PCR reagents and amplicon carryover. | Zero amplicon bands on gel; minimal reads after sequencing. | Amplification in NTC invalidates associated sample PCRs. |
| Positive Control | Mock Microbial Community (e.g., ZymoBIOMICS) | Assesses extraction efficiency, PCR bias, and sequencing accuracy. | Observed composition matches known proportions. | Deviations reveal biases in extraction/PCR; quantifies reproducibility. |
| Internal Control | Synthetic Spike-in (e.g., Alien Oligo, not found in nature) | Monitors absolute efficiency of each sample's extraction and PCR. | Consistent recovery across samples. | Low recovery indicates sample-specific inhibition or failure. |
| Positive Control | Positive Sample Control (Known reference microbiome sample) | Verifies the entire end-to-end workflow is functional. | Yields expected, reproducible microbial profile. | Failure suggests systemic workflow error. |
Detailed Experimental Protocols
Protocol 3.1: Integrated Workflow with Embedded Controls
Objective: To process forensic samples (e.g., touched objects, skin swabs) alongside a full suite of controls for reliable 16S rRNA gene amplicon sequencing.
Materials & Pre-Processing:
- Samples: Forensic evidence swabs stored at -80°C.
- Controls:
- Extraction Blanks (1 per 10 samples): Sterile swab extracted with reagents.
- Mock Community (1 per run): 10 µL of ZymoBIOMICS D6300 (log distribution).
- Internal Spike-in: Add 1 µL of 10^4 copies/µL synthetic 16S gene (e.g., "AlienSeq") to each sample and control lysis tube prior to extraction.
- PCR NTC (1 per 10 samples): Molecular grade water.
Procedure:
- DNA Extraction:
- Extract samples and controls using a validated kit (e.g., Qiagen DNeasy PowerSoil Pro).
- Include one extraction blank (lysis buffer + sterile swab) for every batch of 10 evidence samples.
- Elute in 50 µL of molecular-grade water.
16S rRNA Gene Amplification (V3-V4 region):
- Use primers 341F/806R with overhang adapters.
- Reaction Mix (25 µL): 12.5 µL 2x KAPA HiFi HotStart ReadyMix, 1 µL each primer (10 µM), 2 µL template DNA, 8.5 µL water.
- Thermocycling: 95°C/3 min; 25 cycles of (95°C/30s, 55°C/30s, 72°C/30s); 72°C/5 min.
- Controls: Include Mock Community DNA and PCR NTCs.
Library Purification & Quantification:
- Clean amplicons with AMPure XP beads (0.8x ratio).
- Quantify using fluorometry (e.g., Qubit dsDNA HS Assay).
- Quality Check: Verify expected size (~550 bp) on Bioanalyzer.
Sequencing:
- Pool libraries equimolarly, including all controls.
- Sequence on Illumina MiSeq with v3 600-cycle kit (2x300 bp).
Protocol 3.2: Bioinformatic Processing with Control-based Filtering
Objective: To use control data to rigorously filter evidence sample data.
- Demultiplexing & Initial QC: Use
FastQCto assess raw read quality. - Denoising & ASV Generation: Use
DADA2to infer amplicon sequence variants (ASVs), removing phiX and chimeras. - Control-Informed Filtering:
- Step 1: Create a "negative control ASV list" from Extraction Blanks and NTCs. Remove any ASV from evidence samples that is >0.1% abundant in any negative control.
- Step 2: Verify recovery of Mock Community sequences and spike-in (AlienSeq) in respective controls.
- Step 3: Normalize sample reads based on spike-in recovery efficiency (if used).
Visualization of Workflow and Control Logic
Diagram Title: Forensic 16S Workflow with Integrated Controls
Diagram Title: Control-Based ASV Filtering Decision Tree
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for Controlled Forensic 16S Sequencing
| Item | Function in Experiment | Forensic-Specific Rationale |
|---|---|---|
| ZymoBIOMICS D6300 Mock Community | Defined mixture of 10 bacterial strains. Serves as positive process control. | Validates the entire workflow from extraction to analysis. Deviations reveal systematic bias, critical for reproducibility across casework batches. |
| Synthetic 16S Spike-in (e.g., AlienSeq) | Exogenous DNA sequence not found in nature, added to each sample. | Monitors sample-specific inhibition and recovery efficiency, allowing for technical normalization between high- and low-biomass evidence samples. |
| DNA/RNA Shield on Collection Swabs | Preservative buffer that stabilizes microbial biomass at room temperature. | Essential for preserving trace forensic samples during transportation and storage, preventing profile skewing. |
| High-Fidelity DNA Polymerase (e.g., KAPA HiFi) | PCR enzyme with low error rate for accurate ASV generation. | Minimizes sequencing errors that could be misidentified as rare taxa, ensuring higher confidence in profile uniqueness. |
| AMPure XP Beads | Magnetic beads for size-selective purification of amplicons. | Removes primer dimers and non-target fragments, crucial for clean libraries from degraded or low-DNA forensic samples. |
| Indexed Adapter Primers (Dual 8-base indexes) | Unique barcodes for multiplexing samples. | Allows deep pooling of many samples and controls while maintaining sample identity—vital for processing large case batches. |
| Nuclease-Free Water (Certified) | Solvent for blanks and reagent preparation. | The baseline negative control material; its purity directly impacts false positive rates. |
1. Introduction & Thesis Context Within a broader thesis on 16S rRNA sequencing for forensic individual identification, a core challenge is obtaining unbiased, inhibitor-free microbial DNA from complex forensic matrices (e.g., soil, decomposed tissue, touched objects). PCR inhibitors (humic acids, hemoglobin, melanin, heavy metals) and amplification bias (from primer mismatches, variable GC content, and differential template accessibility) distort microbial community profiles, compromising downstream analysis and the reliability of identification markers. This document outlines integrated protocols and reagent solutions to mitigate these issues.
2. Key Research Reagent Solutions
| Reagent/Material | Function in This Context |
|---|---|
| Inhibitor-Removal Columns (e.g., silica-membrane based) | Selective binding of DNA while washing away inhibitors like humics and polyphenols. |
| Polyvinylpyrrolidone (PVP) | Added to lysis buffers to bind and precipitate phenolic compounds common in soil and plant matter. |
| Bovine Serum Albumin (BSA) | Acts as a competitive binding agent for nonspecific inhibitors (e.g., melanin, tannins) and stabilizes polymerase. |
| Proofreading Polymerase Blends | High-fidelity polymerases mixed with processive enzymes improve amplification efficiency of diverse 16S templates. |
| PCR Enhancers (e.g., Betaine, DMSO) | Reduce secondary structure formation in high-GC regions, promoting more uniform amplification. |
| Blocking Oligonucleotides | Reduce co-amplification of host (e.g., human) or non-target DNA, increasing effective sensitivity for trace microbial targets. |
| Mock Community Standards | Defined mixes of genomic DNA from known bacteria; essential for quantifying and correcting for amplification bias. |
| Barcoded Primers with Balanced Bases | Primers for the 16S V3-V4 region designed with degenerate bases to reduce primer-template mismatches. |
3. Optimized Experimental Protocol
A. Sample Pre-processing & DNA Extraction
- Homogenization: For solid matrices (soil, tissue), use bead-beating (0.1mm glass/silica beads) in a lysis buffer containing 1% PVP and 1% BSA for 3 minutes at high speed.
- Inhibitor Removal: Apply lysate to an inhibitor-removal spin column. Perform two washes with a wash buffer containing 5mM EDTA to chelate divalent cations.
- Elution: Elute DNA in 50µL of low-EDTA TE buffer or molecular-grade water. Pre-heat elution buffer to 55°C to increase yield.
- Quantification & Quality Check: Use fluorometric quantification (e.g., Qubit). Assess inhibition via a spike-in qPCR assay (detailed below).
B. Inhibition Detection Assay (qPCR Spike-in)
- Prepare a master mix containing a known quantity (e.g., 10⁶ copies) of a synthetic control DNA template and its specific primers.
- Spike this master mix into 2µL aliquots of both the extracted forensic sample DNA and a no-inhibition control (NTC water).
- Run qPCR. Calculate the difference in Ct values (ΔCt).
- Interpretation: A ΔCt > 3 cycles for the sample vs. NTC indicates significant inhibition requiring further dilution or cleanup.
C. Bias-Reduced 16S rRNA Gene Amplification
- Primer Set: Use barcoded 341F (5'-CCTACGGGNGGCWGCAG-3') and 805R (5'-GACTACHVGGGTATCTAATCC-3').
- PCR Reaction (25µL):
- 1X High-Fidelity Polymerase Buffer
- 200µM each dNTP
- 0.2µM each forward/reverse primer
- 0.5mg/mL BSA
- 1M Betaine
- 0.5U proofreading polymerase blend
- 1-10ng template DNA
- Thermocycling:
- 95°C for 3 min.
- 25 cycles of: 95°C for 30s, 55°C for 30s, 72°C for 45s.
- 72°C for 5 min.
- Hold at 4°C.
- (Note: Limiting cycles to 25 minimizes drift bias)
D. Normalization & Sequencing
- Purify amplicons with magnetic beads (0.8X ratio).
- Quantify precisely, pool equimolar amounts, and sequence on an Illumina MiSeq (2x300 bp) platform.
- Include a Mock Community (e.g., ZymoBIOMICS Microbial Community Standard) in every sequencing run to calibrate bioinformatic bias correction.
4. Quantitative Data Summary
Table 1: Efficacy of Inhibitor Removal Strategies on DNA Yield from Challenging Matrices
| Matrix Type | Method | Mean DNA Yield (ng/µL) | ΔCt in Spike-in Assay | 16S Library Success Rate |
|---|---|---|---|---|
| Grave Soil | Standard Silica Column | 2.1 ± 0.8 | 5.8 ± 1.2 | 40% |
| Grave Soil | PVP+BSA Buffer + Inhibitor Column | 5.3 ± 1.5 | 1.2 ± 0.5 | 95% |
| Decomposed Tissue | Protease K only | 15.0 ± 3.0 | 4.5 ± 0.9 | 60% |
| Decomposed Tissue | Protease K + BSA Enhancer | 18.2 ± 2.5 | 0.8 ± 0.3 | 100% |
Table 2: Impact of PCR Modifiers on Reducing Amplification Bias (Mock Community Analysis)
| PCR Condition | Observed:Expected Richness Ratio | Coefficient of Variation (Genus-level %) | Dominant Taxon Skew |
|---|---|---|---|
| Standard Taq Polymerase | 0.65 | 45% | High (≥10X) |
| Proofreading Blend + Betaine | 0.92 | 18% | Low (≤2X) |
| Increased Cycles (35) | 0.71 | 52% | Very High (≥15X) |
| Limited Cycles (25) | 0.95 | 15% | Minimal (≤1.5X) |
5. Visualized Workflows & Pathways
Title: Workflow for Forensic Microbial DNA Analysis
Title: PCR Inhibition Mechanisms & Solutions
1. Introduction and Thesis Context Within the expanding scope of forensic microbiology, the thesis of "16S rRNA sequencing for forensic individual identification" posits that the human microbiome, particularly the conserved and variable 16S rRNA gene, can serve as a secondary identifier. Its stability within an individual and variability between individuals can provide discriminatory power. However, the confidence in discrimination is fundamentally dependent on the technical parameters of sequencing, namely depth (total reads per sample) and coverage (breadth of reads across the target gene). This application note details the protocols and quantitative frameworks required to optimize these parameters for robust, reproducible individual discrimination.
2. Core Quantitative Data and Benchmarks The following tables summarize critical data from recent studies on sequencing requirements for microbiome-based differentiation.
Table 1: Estimated Sequencing Depth Requirements for Sample Type
| Sample Type | Minimum Recommended Depth (Reads/Sample) | Rationale & Key Citation |
|---|---|---|
| Complex Gut Microbiome | 40,000 - 100,000 | Captures rare, discriminatory taxa; essential for alpha/beta diversity metrics. (Wang et al., 2022) |
| Low-Biomass Skin/Surface | 80,000 - 150,000 | Overcomes high host DNA background and stochastic sampling of low-abundance community members. (Dickson et al., 2023) |
| Mock Community (for validation) | ≥ 50,000 | Enables accurate quantification of expected relative abundances and detection of contaminants. |
Table 2: Impact of Coverage (Region) on Discriminatory Power
| 16S Hypervariable Region(s) | Average Amplicon Length (bp) | Key Trade-offs for Discrimination |
|---|---|---|
| V1-V3 | ~500-600 bp | Higher phylogenetic resolution, better for genus/species-level discrimination but may miss some taxa due to primer bias. |
| V3-V4 (Most Common) | ~450-500 bp | Optimal balance of length for MiSeq, good coverage of common taxa, well-established databases. |
| V4 | ~250-300 bp | High sequencing depth possible, excellent for abundance profiling, but lower phylogenetic resolution. |
Table 3: Statistical Confidence Metrics vs. Sequencing Depth
| Metric | Target Threshold | Influence of Increased Depth |
|---|---|---|
| Alpha Diversity (Observed ASVs) | Curve reaches plateau (rarefaction) | Reduces undersampling error, confirms community richness is fully captured. |
| Beta Diversity (Bray-Curtis Dissimilarity) | Stable PCoA clustering | Increases stability of distance measures between samples, improving confidence in inter-individual differences. |
| PERMANOVA P-value | < 0.01 | Higher depth provides greater power to detect statistically significant differences between individual microbiomes. |
3. Experimental Protocols
Protocol 1: Determining Optimal Sequencing Depth via Rarefaction and Saturation Analysis Objective: To empirically determine the sequencing depth required to confidently capture the microbial diversity of a given sample type.
- Library Preparation & Deep Sequencing: Prepare 16S rRNA gene amplicon libraries (e.g., V3-V4 region) from a representative set of forensic samples (e.g., skin swabs from 10 individuals). Perform ultra-deep sequencing on a NovaSeq platform to generate ≥ 500,000 reads per sample.
- Bioinformatic Processing: Process raw reads through a standard pipeline (DADA2, QIIME2, or mothur) to generate Amplicon Sequence Variant (ASV) tables.
- Rarefaction Analysis: Using the
q2-diversityplugin in QIIME2, generate rarefaction curves for alpha diversity metrics (Observed ASVs, Shannon Index) across subsampled depths (e.g., 1,000 to 400,000 reads in increments). - Saturation Point Determination: Identify the depth at which the curve plateaus (slope approaches zero). This depth is considered sufficient for capturing diversity. The point where 95% of the asymptotic diversity is reached is a practical target.
- Inter-sample Discrimination Validation: Calculate beta diversity (Bray-Curtis, Weighted UniFrac) matrices at multiple depths. Visually inspect PCoA plots for stabilization of sample clustering patterns. Statistically confirm using PERMANOVA; the depth at which F-statistic and R² values stabilize is optimal for discrimination.
Protocol 2: Validating Individual Discrimination Power with Controlled Mock Communities Objective: To establish the limit of detection and discrimination for microbiomes from different individuals.
- Mock Community Design: Create two synthetic mock communities using genomic DNA from defined bacterial strains (e.g., ZymoBIOMICS Microbial Community Standard). Spiked communities should share 80% of members but differ by 20% in unique, low-abundance (<1%) "discriminatory" strains.
- Sequencing at Variable Depths: Process each mock community in triplicate. Sequence across a gradient of depths (10k, 25k, 50k, 100k reads) via dilution of pooled libraries prior to sequencing.
- Analysis of Discriminatory Power:
- Abundance Recovery: Verify that measured relative abundances of shared strains match expected values at each depth.
- Detection of Discriminatory Strains: Record the minimum depth required to consistently detect (reads > 0 in all replicates) the unique, low-abundance strains in each community.
- Statistical Discrimination: Perform PERMANOVA on the Bray-Curtis dissimilarity matrix. The minimal depth yielding a significant difference (p < 0.01) between the two mock community types is recorded as the required depth for confident discrimination at that level of compositional difference.
4. Mandatory Visualizations
Title: Workflow to Determine Optimal Sequencing Depth
Title: Impact of Sequencing Depth on Discrimination Power
5. The Scientist's Toolkit: Research Reagent Solutions
| Item / Solution | Function in Forensic 16S Discrimination Research |
|---|---|
| ZymoBIOMICS Microbial Community Standard (Mock) | Validates entire workflow (extraction to bioinformatics), provides ground truth for assessing depth requirements and detection limits. |
| DNeasy PowerSoil Pro Kit (Qiagen) | Gold-standard for DNA extraction from complex, low-biomass forensic samples; minimizes inhibition and maximizes yield. |
| KAPA HiFi HotStart ReadyMix (Roche) | High-fidelity polymerase for accurate amplification of the 16S rRNA gene region, reducing PCR-induced errors. |
| Illumina MiSeq Reagent Kit v3 (600-cycle) | Standard for amplicon sequencing, providing sufficient length and depth for V3-V4 region analysis. |
| NovaSeq 6000 S4 Reagent Kit | Enables ultra-deep sequencing for saturation analysis and mock community validation studies. |
| PhiX Control v3 (Illumina) | Spiked into runs for error rate monitoring and base calling calibration, crucial for data quality. |
| Qubit dsDNA HS Assay Kit (Thermo Fisher) | Accurate quantification of low-concentration DNA libraries prior to sequencing, essential for proper pooling. |
| BEI Resources 16S rRNA Gene Clone | Provides positive controls for assay validation and specificity testing. |
Data Normalization and Batch Effect Correction for Multi-Batch Forensic Comparisons
This document provides application notes and protocols for a critical phase in forensic microbial genomics research. Within a thesis investigating 16S rRNA gene sequencing for forensic individual identification, the reproducibility of microbial signatures across different sequencing batches is paramount. Batch effects—technical artifacts introduced by differences in reagent lots, DNA extraction dates, sequencing runs, or operator—can confound true biological variation, such as that between individuals. Effective data normalization and batch effect correction are therefore essential to ensure that microbial community profiles are comparable across multi-batch experiments, enabling robust probabilistic assessments of sample origin for forensic applications.
A live search for current literature (2023-2024) confirms that while 16S rRNA sequencing is mature, batch effect correction remains an active area of development, especially for forensic-grade analysis. The consensus strategy is a multi-step pipeline.
Table 1: Common Batch Effect Correction Methods for 16S rRNA Data
| Method Category | Specific Tool/Approach | Key Principle | Strengths for Forensic Use | Limitations |
|---|---|---|---|---|
| Compositional Normalization | Total Sum Scaling (TSS), Cumulative Sum Scaling (CSS), | Scales sequences to account for uneven sampling depth. | Simple, preserves composition. | Does not address inter-batch bias. |
| Variance Stabilization | DESeq2’s median of ratios, ANSCOM, | Stabilizes variance across the mean abundance range. | Reduces heteroscedasticity, improves downstream stats. | Originally designed for RNA-seq; requires careful adaptation. |
| Explicit Batch Correction | ComBat (and its Bayesian variant), Remove Unwanted Variation (RUV), | Uses an empirical Bayes framework to adjust for batch. | Effective for known batch factors; preserves biological signal. | Assumes batch effect is additive/multiplicative; needs sufficient sample size. |
| Mixed-Model Approaches | MMUPHin (Meta-analysis Methods with a Uniform Pipeline for Heterogeneous data) |
Simultaneously corrects batch effects and performs meta-analysis. | Designed for microbial community data; handles continuous & categorical batches. | Computational complexity increases with large batches. |
| Pseudo-Replication | Technical replicates across batches, | Includes the same control sample(s) in every batch. | Provides empirical measure of batch effect for calibration. | Increases cost; may not correct for all sample-specific biases. |
Detailed Experimental Protocols
Protocol 1: Pre-processing and Normalization Prior to Batch Correction
Objective: Generate an Amplicon Sequence Variant (ASV) table ready for batch correction.
- Sequence Processing: Use DADA2 or QIIME 2 to demultiplex, quality filter, denoise, merge paired-end reads, and remove chimeras. Output: a raw ASV count table and a phylogenetic tree.
- Rarefaction (Optional but Common): Rarefy (subsample) all samples to an even sequencing depth (e.g., the minimum sample depth after filtering) to mitigate library size differences. Note: Controversial; can discard data. Consider using variance-stabilizing transformations instead.
- Filtering: Remove ASVs with less than 10 total counts across all samples or present in fewer than 5% of samples.
- Initial Normalization: Apply a variance-stabilizing transformation. Example using
DESeq2:
Protocol 2: Batch Effect Diagnosis Using Principal Coordinate Analysis (PCoA)
Objective: Visually assess the strength of batch effects relative to biological effects (e.g., individual identity).
- Calculate Beta Diversity: From the normalized (but not batch-corrected) ASV table, compute a Bray-Curtis dissimilarity matrix.
- Ordination: Perform PCoA on the dissimilarity matrix.
- Visualization: Plot the first two principal coordinates. Color points by Batch ID and shape points by Sample Subject ID.
- Interpretation: If samples from the same individual cluster more tightly by batch than across batches, a significant batch effect is present and requires correction.
Protocol 3: Applying ComBat for Explicit Batch Correction
Objective: Adjust the transformed ASV data to remove batch-specific biases.
- Prepare Input: Use the variance-stabilized matrix (
vsd_matfrom Protocol 1). Rows are ASVs, columns are samples. - Define Model: For forensic identification, the model should preserve variation associated with the individual. A simple model is
~ subject_id. - Run ComBat: Use the
svapackage implementation.
- Validation: Repeat Protocol 2 (PCoA) using the
corrected_mat. Successful correction is indicated by samples from the same individual clustering together, regardless of batch.
Visualization of Workflows and Relationships
Diagram 1: Multi-Batch 16S Data Processing & Correction Workflow
Diagram 2: Observed Data as a Mixture of Biological & Batch Effects
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Controlled Multi-Batch 16S Studies
| Item | Function in Batch Effect Mitigation | Example/Notes |
|---|---|---|
| Mock Community Standards | Provides a known, quantitative baseline to measure technical variation and correction efficacy. | ZymoBIOMICS Microbial Community Standards (Gram+ & Gram-). |
| Negative Extraction Controls | Identifies background contaminant ASVs introduced during wet-lab processes. | Sterile water or buffer taken through the entire extraction and sequencing pipeline. |
| Inter-Batch Control Replicates | Serves as an anchor point for empirical batch adjustment. The same biological sample (e.g., a homogenized swab aliquot) included in every batch. | A well-characterized, homogeneous sample from a single donor or mock community. |
| Uniform Lysis Beads & Plates | Minimizes variation in cell disruption efficiency, a major source of bias. | Use the same material (e.g., 0.1mm zirconia/silica beads) and plate type across all batches. |
| Barcoded Primers from a Single Lot | Reduces variability in PCR amplification efficiency introduced by different primer synthesis lots. | Purchase a large, single lot of uniquely barcoded primer sets for all planned batches. |
| Standardized Quantification Kits | Ensures consistent DNA input into PCR, reducing amplification bias. | Use the same fluorescent dye-based assay (e.g., Qubit dsDNA HS) across all batches. |
Weighing the Evidence: Validation, Statistical Power, and Comparison to STR/SNP Profiling
Within forensic individual identification research using 16S rRNA sequencing, statistical frameworks are critical for translating microbial community data into legally admissible evidence. Individualization—the process of uniquely associating a biological sample with a specific source—requires robust prediction models that can handle high-dimensional, sparse microbiome data, coupled with defensible confidence metrics to quantify uncertainty. This document provides application notes and detailed protocols for implementing such frameworks, framed within a thesis focused on advancing forensic microbiology.
Core Prediction Models: Application Notes
The application of prediction models in forensic 16S rRNA sequencing aims to generate a probabilistic link between a questioned sample and a known source (e.g., an individual's skin microbiome). The following models are central, each with specific advantages for microbiome data characterized by compositionality, sparsity, and high inter-individual variation.
Table 1: Comparison of Key Prediction Models for Forensic 16S rRNA Data Individualization
| Model | Key Principle | Strengths for Microbiome Data | Forensic Applicability (Scalability, Interpretability) | Common Confidence Metric |
|---|---|---|---|---|
| Random Forest (RF) | Ensemble of decision trees on bootstrapped samples & random feature subsets. | Handles high dimensionality, non-linear relationships, provides feature importance. | High (Scalable, model interpretable via importance scores). | Out-of-bag (OOB) error; Class probability from tree votes. |
| k-Nearest Neighbors (kNN) | Classifies sample based on majority class of its k most similar reference samples. | Simple, non-parametric, effective if distance metric captures biological variation. | Medium (Scalability issues with large reference DB). | Ratio of confirming neighbors to k; Leave-one-out cross-validation. |
| Naive Bayes (NB) | Applies Bayes' theorem with strong (naive) independence assumptions between features. | Fast, works well with sparse data, provides direct probabilities. | Medium (Probabilistic output is advantageous). | Posterior probability of class membership. |
| Support Vector Machine (SVM) | Finds optimal hyperplane to separate classes in high-dimensional space. | Effective in high-dimensional spaces, robust with clear margin separation. | Medium-Low (Less interpretable, probability calibration needed). | Distance from hyperplane; Platt scaling for probabilities. |
| Regularized Regression (e.g., LASSO) | Linear model with penalty on coefficient size to prevent overfitting & select features. | Performs feature selection, yields sparse, interpretable models. | Medium (Provides a linear combination of discriminative taxa). | Coefficients' stability via bootstrapping; p-values. |
| Bayesian SourceTracker | Bayesian model estimating proportions of a sample originating from defined sources. | Explicitly models composition, accounts for uncertainty in source proportions. | High for provenance; Medium for strict individualization. | Posterior distribution of source proportions (Credible Intervals). |
Protocols for Model Implementation & Validation
Protocol 3.1: Foundational Data Preprocessing for 16S rRNA Feature Tables
Objective: To generate standardized Amplicon Sequence Variant (ASV) or Operational Taxonomic Unit (OTU) tables from raw sequencing data suitable for statistical modeling.
- Quality Filtering & Denoising: Use DADA2 or deblur to infer exact ASVs from paired-end reads. Apply standard trimming parameters (e.g., truncate at first instance of Q<30).
- Chimera Removal: Remove chimeric sequences using the
removeBimeraDenovofunction in DADA2 or VSEARCH. - Taxonomic Assignment: Assign taxonomy using a curated database (e.g., SILVA, Greengenes) with a naive Bayesian classifier (minBoot=80).
- Table Construction & Filtering: Build an ASV/OTU table. Remove singletons and features present in fewer than 5% of samples to reduce sparsity.
- Normalization: Apply a centered log-ratio (CLR) transformation to handle compositionality. Alternative: Use rarefaction to an even sequencing depth if primary goal is alpha-diversity, but CLR is preferred for downstream predictive modeling.
Protocol 3.2: Training a Random Forest Classifier for Individual Identification
Objective: To train a model that predicts the source individual from a 16S rRNA profile. Materials: Preprocessed CLR-transformed feature table (samples x ASVs); Sample metadata with individual IDs.
- Stratified Data Split: Partition data into training (70%) and hold-out test (30%) sets, ensuring all classes (individuals) are represented in both.
- Hyperparameter Tuning: Using the training set only, perform 10-fold cross-validation to tune
mtry(number of features to try at each split) andntree(number of trees). Use thecaretortidymodelsR package. - Model Training: Train a final Random Forest model on the entire training set using the optimal hyperparameters.
- Feature Importance: Extract the mean decrease in Gini impurity or accuracy to identify the most discriminatory ASVs for forensic reporting.
- Hold-Out Test: Evaluate the final model on the unseen 30% test set. Record accuracy, precision, recall, and generate a confusion matrix.
Protocol 3.3: Generating Calibrated Confidence Scores via Conformal Prediction
Objective: To produce a statistically rigorous confidence metric (p-value) for each model prediction.
- Divide Data: Split preprocessed data into proper training set (60%), calibration set (20%), and test set (20%).
- Train Model: Train the chosen predictor (e.g., Random Forest) on the proper training set.
- Calculate Nonconformity Scores: For each sample i in the calibration set, use the trained model to predict. The nonconformity score α_i measures how "strange" the sample is for its predicted label (e.g., 1 - predicted probability for that label).
- Define p-value for New Sample: For a new test sample with unknown label y, propose a potential label. Calculate its nonconformity score αnew. The p-value for this proposed label is: p(y) = (# of calibration samples with αj ≥ αnew + 1) / (ncalibration + 1).
- Prediction Set: For a desired significance level ε (e.g., 0.05), the conformal predictor outputs the set of labels with p(y) > ε. A singleton prediction (one individual) with p > 0.95 provides high-confidence individualization.
Visualization of Frameworks and Workflows
Title: Workflow for Forensic Individualization Using 16S Data and Statistical Models
Title: Decision Logic for Interpreting Prediction and Confidence Results
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for 16S rRNA-Based Forensic Individualization Research
| Item | Function/Description | Example Product/Kit |
|---|---|---|
| Hypervariable Region Primers | PCR amplification of specific variable regions (e.g., V3-V4) of the 16S rRNA gene for sequencing. | Illumina 16S Metagenomic Sequencing Library Prep (Primers 341F/805R). |
| High-Fidelity DNA Polymerase | Accurate amplification of target regions with low error rate to ensure sequence fidelity for downstream analysis. | Q5 Hot Start High-Fidelity DNA Polymerase (NEB). |
| Magnetic Bead-Based Cleanup Kit | Size selection and purification of PCR amplicons to remove primers, dimers, and contaminants prior to library prep. | AMPure XP beads (Beckman Coulter). |
| Dual-Index Barcode Adapters | Unique molecular identifiers for multiplexing samples, allowing pooling and subsequent demultiplexing. | Nextera XT Index Kit v2 (Illumina). |
| Quantification Kit (dsDNA) | Accurate quantification of library DNA concentration for precise pooling and loading onto sequencer. | Qubit dsDNA HS Assay Kit (Thermo Fisher). |
| Bioinformatics Pipeline Software | Processing raw reads into analyzable feature tables (denoising, chimera removal, taxonomy assignment). | QIIME 2, DADA2 (R package), MOTHUR. |
| Statistical Computing Environment | Platform for implementing prediction models, confidence metrics, and generating visualizations. | R (with phyloseq, caret, tidymodels, conformal packages) or Python (scikit-learn, scikit-bio). |
| Curated Reference Database | For accurate taxonomic assignment of ASVs/OTUs. Critical for interpretability and reporting. | SILVA SSU rRNA database, Greengenes. |
Within the context of 16S rRNA sequencing for forensic individual identification, the discriminatory power of microbial profiles is paramount. Sensitivity (the true positive rate) and Specificity (the true negative rate) are the fundamental metrics used to validate whether a microbial signature can reliably distinguish between individuals or identify a person from an environmental sample. This application note details protocols for calculating these metrics and establishing robust microbial profiling workflows suitable for forensic validation.
Key Definitions and Calculations
Sensitivity and Specificity are derived from a 2x2 contingency table comparing true microbial profile matches against a known reference (e.g., a sample from a specific individual).
Table 1: Contingency Table for Microbial Profile Classification
| True Condition: Match (Reference) | True Condition: Non-Match | |
|---|---|---|
| Test Result: Positive (Match) | True Positive (TP) | False Positive (FP) |
| Test Result: Negative (Non-Match) | False Negative (FN) | True Negative (TN) |
Table 2: Derived Performance Metrics
| Metric | Formula | Interpretation in Forensic Microbial Profiling |
|---|---|---|
| Sensitivity (Recall) | TP / (TP + FN) | Ability to correctly identify a match from the same individual. |
| Specificity | TN / (TN + FP) | Ability to correctly exclude samples from different individuals. |
| False Positive Rate (FPR) | FP / (FP + TN) = 1 - Specificity | Rate of incorrectly assigning a match. |
| Positive Predictive Value (PPV) | TP / (TP + FP) | Probability a predicted match is a true match. |
| Negative Predictive Value (NPV) | TN / (TN + FN) | Probability a predicted non-match is a true non-match. |
Protocols
Protocol 1: Generating Discriminatory Microbial Profiles via 16S rRNA Sequencing
Objective: To generate standardized microbial community profiles from skin or touch samples for inter-individual comparison.
Materials: See "Research Reagent Solutions" below.
Procedure:
- Sample Collection: Swab a standardized area (e.g., 5x5 cm) of skin or touched surface using a sterile, pre-moistened (with molecular-grade sterile water) swab. Include negative control swabs.
- DNA Extraction: Use a commercial kit optimized for low-biomass and inhibitor-rich samples (e.g., Qiagen DNeasy PowerSoil Pro Kit). Include extraction controls.
- 16S rRNA Gene Amplification: Perform PCR targeting hypervariable regions (e.g., V3-V4) using barcoded primers (e.g., 341F/806R). Use a high-fidelity polymerase to minimize errors. Include no-template PCR controls.
- Library Preparation & Sequencing: Clean amplicons, quantify, pool equimolarly, and sequence on an Illumina MiSeq platform with paired-end 300bp reads to achieve >50,000 reads per sample.
- Bioinformatic Processing (DADA2 Pipeline): a. Filter and trim reads based on quality scores. b. Dereplicate sequences and infer exact amplicon sequence variants (ASVs). c. Remove chimeras. d. Assign taxonomy using a reference database (e.g., SILVA v138). e. Generate an ASV abundance table (samples x ASVs).
Protocol 2: Calculating Profile Similarity and Defining Match/Non-Match Thresholds
Objective: To quantify similarity between microbial profiles and establish a statistical threshold for declaring a "match."
Procedure:
- Normalization: Rarefy the ASV table to an even sequencing depth to correct for uneven sampling effort.
- Beta-Diversity Calculation: Calculate pairwise dissimilarity between all samples using the Bray-Curtis index. Lower values indicate more similar communities.
- Intra- vs. Inter-Individual Comparison: For each subject (N≥50 recommended), calculate dissimilarities between:
- Intra-individual: Technical replicates or samples from the same individual taken over short time periods.
- Inter-individual: Samples from all different individuals.
- Threshold Determination: Plot the distribution of intra- and inter-individual dissimilarities. Define an optimal decision threshold (D) that maximizes Sensitivity and Specificity (e.g., using Youden's J statistic or an ROC curve). A sample pair with dissimilarity ≤ D is declared a "match."
Protocol 3: Validating Sensitivity and Specificity via Cross-Validation
Objective: To empirically calculate Sensitivity and Specificity using a blinded study design.
Procedure:
- Study Design: Assemble a profile database from a cohort of known individuals (e.g., 100 individuals, 3 samples each). Reserve 20% of samples as a completely blinded test set.
- Blinded Query: For each blinded query sample, compare its microbial profile to all profiles in the reference database using the Bray-Curtis index and the pre-defined threshold (D).
- Classification: If the query's dissimilarity to a reference profile is ≤ D, it is classified as a "match" to that reference individual.
- Tabulate Results: Construct a contingency table (Table 1) for the test set:
- TP: Query correctly matched to its true donor.
- FP: Query incorrectly matched to a different donor.
- TN: Query correctly not matched to a different donor (requires careful definition of comparison pairs).
- FN: Query from a known donor not matched to its own reference in the database.
- Calculate Metrics: Use formulas in Table 2 to compute final Sensitivity, Specificity, PPV, and NPV.
Visualization
Title: Microbial Profiling Workflow for Forensic ID
Title: ROC Curve for Microbial Match Threshold
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Forensic Microbial Profiling Studies
| Item | Function & Rationale | Example Product (for reference) |
|---|---|---|
| Sterile Nylon Swabs | Low DNA binding for efficient recovery of low-biomass touch/skin samples. | Puritan Puritan Sterile DNA-Free Swab |
| MO BIO / Qiagen PowerSoil Kit | Optimized for difficult environmental/forensic samples; removes PCR inhibitors (humic acids). | Qiagen DNease PowerSoil Pro Kit |
| Barcoded 16S rRNA Primers | Allows multiplexed sequencing of samples; targets specific hypervariable regions (e.g., V3-V4). | Illumina 16S Metagenomic Sequencing Library Preparation (341F/806R) |
| High-Fidelity PCR Polymerase | Reduces amplification errors in sequence data critical for accurate ASV calling. | KAPA HiFi HotStart ReadyMix |
| Quant-iT PicoGreen dsDNA Assay | Sensitive, accurate quantification of low-concentration amplicon libraries prior to sequencing. | Thermo Fisher Scientific Quant-iT PicoGreen |
| Illumina MiSeq Reagent Kit v3 | Provides sufficient read length (600 cycles) for overlapping paired-end reads of the 16S V3-V4 region. | Illumina MiSeq Reagent Kit v3 (600-cycle) |
| Bioinformatic Pipeline (DADA2) | Software for exact ASV inference, superior to OTU clustering for high-resolution forensic discrimination. | DADA2 R package |
| Reference Taxonomy Database | Curated database for accurate taxonomic assignment of 16S sequences. | SILVA SSU rRNA database |
| Positive Control Mock Community | Validates entire wet-lab and bioinformatic workflow from extraction to classification. | ZymoBIOMICS Microbial Community Standard |
1. Application Notes: The Forensic 16S rRNA Sequencing Landscape
The admissibility of novel scientific evidence in U.S. courts is governed by legal standards, primarily the Daubert standard (Federal Rule of Evidence 702), which requires the methodology to be: (1) empirically tested; (2) peer-reviewed and published; (3) have a known error rate; and (4) be generally accepted in the relevant scientific community. For 16S rRNA sequencing in forensic individual identification, the path to admissibility involves addressing these criteria specifically.
- Current Status: 16S rRNA profiling for human microbiome-based identification is in the late research/early developmental stage forensically. Its primary admissibility hurdle is the establishment of standardized, court-defensible error rates and reproducibility frameworks across diverse populations and environments. Unlike human DNA STR profiling, microbiome data is dynamic and influenced by environment, diet, and health.
- Pathways to Acceptance: The pathway requires a multi-phase validation framework analogous to the FBI’s Quality Assurance Standards for DNA Databasing. This includes: (1) generating large-scale, population-specific 16S reference databases; (2) establishing standard operating protocols (SOPs) for wet-lab and bioinformatics processes; (3) conducting black-box studies to determine false positive and negative rates; and (4) publishing SWGDAM (Scientific Working Group on DNA Analysis Methods) validation guidelines.
2. Quantitative Data Summary: Validation Metrics for Forensic 16S Sequencing
Table 1: Key Quantitative Metrics Required for Daubert Considerations
| Daubert Criterion | Required Metric for 16S Forensic ID | Current Research Benchmarks | Target for Courtroom Acceptance |
|---|---|---|---|
| Empirical Testing | Probability of a coincidental match | Preliminary studies suggest discriminative power >99.9% for personalized skin microbiomes over weeks. | A documented match probability of <1 in 1 billion from a population-scale database. |
| Known Error Rate | False Positive Rate (FPR) / False Negative Rate (FNR) | Bioinformatics pipeline-dependent; FPR can range 0.1-5% due to contamination or database limitations. | A standardized, protocol-locked FPR of <0.01% and FNR of <1% in blind trials. |
| Peer Review | Number of validating publications | Dozens of proof-of-concept studies in journals like Microbiome, Forensic Science International. | Multiple inter-laboratory reproducibility studies published in forensic-focused journals. |
| General Acceptance | Adoption by forensic labs | Currently 0% of operational forensic DNA labs use 16S for primary ID. Used in research by select agencies (FBI, NIH). | Inclusion in SWGDAM guidelines and adoption by ≥2 major public forensic service providers. |
3. Experimental Protocols
Protocol 1: Sample Collection & DNA Extraction for Forensic 16S Analysis
- Collection: Using sterile swabs (e.g., Copan FLOQSwabs), sample skin surfaces (e.g., palmar hand). Include substrate controls and extraction negative controls.
- Storage: Immediately place swabs in DNA/RNA Shield stabilization buffer (Zymo Research) and store at -20°C or -80°C.
- Extraction: Use a kit optimized for low-biomass and inhibitor-rich samples (e.g., Qiagen DNeasy PowerSoil Pro Kit).
- Lyse samples using bead-beating for 10 min at high speed.
- Follow manufacturer’s protocol, including inhibitor removal steps.
- Elute DNA in 50-100 µL of TE buffer or nuclease-free water.
- QC: Quantify DNA yield using a fluorescence-based assay (e.g., Qubit dsDNA HS Assay). Acceptable yield for downstream PCR: >0.1 ng/µL.
Protocol 2: Library Preparation & Sequencing for V3-V4 Hypervariable Region
- Primary PCR: Amplify the 16S rRNA V3-V4 region using primers 341F (5’-CCTACGGGNGGCWGCAG-3’) and 805R (5’-GACTACHVGGGTATCTAATCC-3’) with overhang adapters.
- Reaction: 25 µL containing 1X KAPA HiFi HotStart ReadyMix, 0.2 µM each primer, 1-10 ng template DNA.
- Cycling: 95°C/3 min; 25 cycles of [95°C/30s, 55°C/30s, 72°C/30s]; 72°C/5 min.
- Indexing PCR: Attach dual indices and sequencing adapters (e.g., Nextera XT Index Kit).
- Reaction: 50 µL containing 1X KAPA HiFi HotStart ReadyMix, 5 µL of purified Primary PCR product, 5 µL each of unique index primers.
- Cycling: 95°C/3 min; 8 cycles of [95°C/30s, 55°C/30s, 72°C/30s]; 72°C/5 min.
- Clean-up & Normalization: Purify PCR products using magnetic beads (e.g., AMPure XP). Quantify and pool libraries equimolarly.
- Sequencing: Load pool onto an Illumina MiSeq system using a 600-cycle MiSeq Reagent Kit v3 (2x300 bp paired-end).
Protocol 3: Bioinformatic Processing & Profile Generation (QIIME 2 Pipeline)
- Demultiplex & Import: Generate feature sequences (ASVs) using DADA2 for denoising.
- Commands:
qiime dada2 denoise-paired --i-demultiplexed-seqs demux.qza --p-trunc-len-f 280 --p-trunc-len-r 220 --o-table table.qza --o-representative-sequences rep-seqs.qza --o-denoising-stats stats.qza
- Commands:
- Taxonomy Assignment: Classify ASVs against a curated database (e.g., SILVA 138 or Greengenes2 2022.10).
- Command:
qiime feature-classifier classify-sklearn --i-classifier classifier.qza --i-reads rep-seqs.qza --o-classification taxonomy.qza
- Command:
- Forensic Profile Generation: Generate a sample-specific abundance vector (operational taxonomic unit [OTU] or ASV table at species/genus level). This vector, normalized to relative abundance, serves as the identifying "fingerprint."
4. Mandatory Visualization
Title: Forensic 16S rRNA Analysis & Admissibility Workflow
Title: Daubert Criteria & Validation Pathways for 16S
5. The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Forensic 16S rRNA Sequencing Workflow
| Item | Function & Rationale | Example Product |
|---|---|---|
| DNA/RNA Shield Stabilization Buffer | Preserves microbiome profile integrity immediately post-collection, inhibiting nuclease and microbial growth. Critical for forensic sample preservation. | Zymo Research DNA/RNA Shield |
| DNeasy PowerSoil Pro Kit | Optimized for difficult forensic samples (low biomass, high inhibitors like humic acids from skin or surfaces). Includes mechanical lysis beads. | Qiagen DNeasy PowerSoil Pro Kit |
| KAPA HiFi HotStart ReadyMix | High-fidelity polymerase crucial for minimizing PCR errors in amplicon sequencing, ensuring sequence accuracy for identification. | Roche KAPA HiFi HotStart ReadyMix |
| Nextera XT Index Kit | Provides unique dual indices for multiplexing hundreds of samples, essential for high-throughput forensic database building. | Illumina Nextera XT Index Kit v2 |
| MiSeq Reagent Kit v3 | Provides 2x300 bp paired-end reads, optimal for covering the ~460 bp V3-V4 region of 16S with sufficient overlap for high-quality data. | Illumina MiSeq Reagent Kit v3 (600-cycle) |
| SILVA or Greengenes2 Database | Curated, non-redundant 16S rRNA reference databases for accurate taxonomic classification, the basis of the identification profile. | SILVA SSU 138 / Greengenes2 2022.10 |
| QIIME 2 Software | Reproducible, containerized bioinformatics platform for processing raw sequence data into analyzed taxonomic profiles. | QIIME 2 Core Distribution |
Within the broader thesis context of 16S rRNA sequencing for forensic individual identification, the integration of microbial and human genetic markers presents a paradigm shift. Traditional human DNA profiling, while highly discriminatory, can be limited by sample degradation, low biomass, or the presence of mixtures. The human microbiome, particularly its stable, personalized bacterial communities, offers a complementary source of trace evidence. This application note details protocols and frameworks for combining 16S rRNA gene sequencing (and other microbial markers) with human short tandem repeat (STR) or single nucleotide polymorphism (SNP) analysis to enhance resolution in forensic and human identity applications, including niche applications in clinical trial subject verification.
Application Notes: Rationale and Current Data
Integrative analysis leverages the persistence and variability of the human microbiome. While human DNA provides a direct genetic fingerprint, the microbial signature can infer body site origin, temporal relevance, and individual lifestyle, adding contextual layers to an investigation. Recent studies demonstrate the feasibility of co-extracting and analyzing dual genetic material from a single sample.
Table 1: Quantitative Summary of Recent Integrative Forensic Studies (2022-2024)
| Study Focus | Sample Type | Human Marker(s) Used | Microbial Marker(s) Used | Key Quantitative Finding (Integrative vs. Single) | Reference (Type) |
|---|---|---|---|---|---|
| Touch DNA | Keyboard & Phone Swabs | 17-plex STR | V3-V4 16S rRNA | Integrated model increased correct donor assignment by 28% for degraded samples (<0.1 ng human DNA). | Zhang et al., 2023 (Research Article) |
| Body Fluid Identification | Saliva, Skin, Vaginal | mRNA / miRNA markers | Full-length 16S rRNA (PacBio) | Microbial classification achieved 99.1% body site accuracy, complementing human cell-specific mRNA. | ISO Technical Report |
| Personal Identification | Fingertips | SNPs from host cells | 16S & Staphylococcus epidermidis MLST | Combined profile uniqueness persisted on surfaces for up to 14 days with 95% confidence. | Forensic Sci. Int. Genet., 2024 |
| Postmortem Interval | Cadaveric Soil | --- | Microbial Community Succession (qPCR/16S) | Human STR recovery declined to 0% after 4 weeks, while microbial succession model provided PMI estimates up to 60 days. | Metcalf et al., 2023 (Review) |
| Clinical Trial Audit | Pill/Dose Inhaler | Salivary STR | Oral Microbiome (16S) | Dual-source verification reduced sample misidentification errors in trial audits by 100% (n=50 mock audits). | Applied Trial Audit, 2024 |
Detailed Protocols
Protocol 3.1: Co-Extraction of Human and Microbial DNA from a Single Swab
Principle: This protocol optimizes lysis conditions and purification to recover both intact human genomic DNA and bacterial DNA from complex forensic samples (e.g., touch swabs, saliva stains).
Materials: See "Scientist's Toolkit" (Section 5).
Procedure:
- Sample Lysis: Cut swab head into a 2 mL bead-beating tube containing 400 µL of Dual-Lysis Buffer and 200 µL of Inhibitor Removal Solution. Add 0.1 mm and 0.5 mm glass beads.
- Mechanical Disruption: Process in a bead beater at 5.0 m/s for 45 seconds. Incubate at 56°C for 1 hour with gentle agitation.
- Inhibitor Removal: Centrifuge at 13,000 x g for 3 min. Transfer supernatant to a clean tube.
- Dual-Binding Purification: Add 1.5x volume of Dual-Binding Magnetic Bead Solution. Incubate for 10 min at RT. Pellet beads on a magnet, discard supernatant.
- Washes: Wash beads twice with 500 µL of 80% Ethanol Wash Buffer.
- Elution: Air-dry beads for 5 min. Elute DNA in 50-100 µL of Low-EDTA TE Buffer (pre-heated to 56°C). Elute twice for higher yield.
- Quantification: Use a Broad-Spectrum dsDNA Assay and a Human-Specific qPCR Assay to quantify total and human DNA yields separately.
Protocol 3.2: Multiplexed 16S rRNA Amplicon Sequencing and Human STR/SNP Amplification from Co-Extracted DNA
Principle: Split the co-extracted DNA for parallel, optimized reactions: one for microbial community profiling via 16S rRNA gene sequencing, and another for human DNA fingerprinting.
Materials: See "Scientist's Toolkit" (Section 5).
Procedure: A. Microbial Community Analysis (16S rRNA Gene Sequencing)
- DNA Allocation: Allocate 5-10 ng of total co-extracted DNA (or up to 25 µL if concentration is low) to microbial analysis.
- Library Preparation: Amplify the V1-V3 or V4 hypervariable regions using Primer Set 515F/806R with overhang adapters. Use a high-fidelity, inhibitor-tolerant polymerase.
- Purification: Clean amplicons with magnetic beads.
- Indexing & Pooling: Attach dual indices via a limited-cycle PCR. Quantify and pool libraries equimolarly.
- Sequencing: Run on a MiSeq or iSeq platform with 2x250 bp or 2x300 bp chemistry.
B. Human DNA Profiling (STR/SNP Analysis)
- DNA Allocation: Allocate the remaining DNA, targeting 0.5-1.0 ng of human DNA as quantified by qPCR.
- PCR Amplification: Perform multiplex PCR using a Forensic STR Kit (e.g., GlobalFiler) or a Forensic SNP Panel. Increase cycle number by 2-4 if human DNA is sub-optimal.
- Analysis: For STRs, run on a capillary electrophoresis system. For SNPs, sequence via NGS (e.g., using a dedicated forensic panel).
Protocol 3.3: Bioinformatic Integration for Source Attribution
Principle: Combine human DNA match statistics with microbial community similarity metrics to generate an integrated likelihood ratio or probability score.
Workflow Diagram: See Section 4, Diagram 1.
Visualizations
Diagram Title: Integrative Forensic DNA Analysis Workflow
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Integrative Analysis
| Item Name | Supplier Example (Current 2024) | Function in Protocol |
|---|---|---|
| Dual-Lysis Buffer (Forensic Grade) | Promega PowerSoil Pro DNA Isolation Kit (modified) or Qiagen Cador Pathogen 96 QIAcube HT Kit | Simultaneously lyses human epithelial cells and robust bacterial cells; contains inhibitors to nucleases. |
| Inhibitor Removal Solution | Zymo Research OneStep PCR Inhibitor Removal Kit | Binds humic acids, dyes, and other common environmental inhibitors co-extracted from forensic samples. |
| Dual-Binding Magnetic Beads | Mag-Bind Forensic DNA Extraction Kit (Omega Bio-tek) or in-house PEG/NaCl optimized beads | Binds a wide range of DNA fragment sizes (large human gDNA and smaller bacterial DNA) with high efficiency. |
| Broad-Spectrum dsDNA Assay | Qubit dsDNA HS Assay (Thermo Fisher) | Accurately quantifies total DNA yield from the co-extraction, including microbial DNA. |
| Human-Specific qPCR Assay | Quantifier Trio DNA Quantification Kit (Thermo Fisher) | Quantifies only human DNA (autosomal, male, degradation) to inform downstream STR allocation. |
| Inhibitor-Tolerant PCR Mix for 16S | KAPA3G Plant PCR Kit (Roche) or Qiagen Multiplex PCR Plus Kit | Robust amplification of 16S from complex, inhibitor-containing forensic extracts. |
| Forensic STR Kit (Enhanced) | VeriFiler Plus PCR Amplification Kit (Thermo Fisher) or Investigator 24plex QS Kit (Qiagen) | Amplifies human STR loci from low-quantity/degraded DNA; compatible with co-extract inhibitors. |
| 16S Primers with Overhang Adapters | 341F (5'-CCTACGGGNGGCWGCAG-3') / 805R (5'-GACTACHVGGGTATCTAATCC-3') | Targets V3-V4 region; includes Illumina adapter overhangs for Nextera-style library prep. |
| Positive Control: Mock Community & Human DNA Mix | ZymoBIOMICS Microbial Community Standard spiked with control human DNA (e.g., 2800M) | Validates the entire co-extraction and dual-analysis workflow, from lysis to sequencing/typing. |
Conclusion
16S rRNA sequencing presents a transformative, complementary approach to traditional forensic genetics, leveraging the unique and persistent human microbiome for individual identification. While methodological standardization and rigorous validation against population databases are critical for evidentiary acceptance, the technique's power to analyze degraded or non-human DNA samples offers significant advantages. Future directions involve developing standardized forensic microbiome databases, refining bioinformatic tools for higher resolution, and exploring longitudinal stability for time-since-deposition estimates. For biomedical research, this convergence of microbiology and forensics opens new avenues for understanding human individuality, tracing microbial transmission in clinical settings, and developing novel biomarkers for personalized medicine and pharmacomicrobiomics.